α,ω-链端功能化聚烯烃的设计与合成新进展
2016-08-02天津科技大学化工与材料学院天津300457中国科学院上海有机化学研究所上海0003
姜 涛,许 芳,,董 鹏,,马 志(. 天津科技大学化工与材料学院,天津 300457;. 中国科学院上海有机化学研究所,上海 0003)
α,ω-链端功能化聚烯烃的设计与合成新进展
姜 涛1,许 芳1,2,董 鹏1,2,马 志2
(1. 天津科技大学化工与材料学院,天津 300457;2. 中国科学院上海有机化学研究所,上海 200032)
链端功能化聚烯烃的设计与合成是近些年来的研究热点之一.合成α,ω-链端功能化聚烯烃的方法较多,活性聚合一般需要适当的金属催化剂,得到的聚合物相对分子质量分布窄,而且单体消耗完以后聚合物链仍保持活性,加入新的单体以后增长可以继续进行,以此来制备嵌段共聚物,也可以加入终止剂终止聚合反应制备双端功能化聚烯烃.叶立德同源聚合通过引入带有不同功能基团的硼烷与叶立德进行反应,得到不同反应性基团封端的聚烯烃,再经过进一步的基团转化反应,可以得到多种不同的端基功能化的聚烯烃.此外,烯烃聚合过程中向链转移剂进行链转移反应、开环易位聚合以及与点击化学结合都可以用来制备链端功能化的聚烯烃.链端功能化聚烯烃不仅可以用作聚合物改性剂,而且更是构筑其他更复杂结构大分子的出发点,例如嵌段共聚物、接枝共聚物以及更为复杂的聚合物分子刷.
聚烯烃;α,ω-链端功能化;链转移反应;活性聚合;叶立德同源聚合;“Click”反应
人类利用天然聚合物的历史久远,但是直到19世纪中叶才开始对天然聚合物的化学改性工作.现在我们生活中所用到的高分子材料都是人工合成的,其中很多应用都与材料的物理化学性能有关,但是大多数高分子材料不能同时满足实际应用对它的要求,因此常需要针对不同的性能(如润湿性、生物相容性、印刷性能等)对其进行改性处理.对高分子材料进行改性的方法有很多,包括共混改性、化学改性、表面改性等诸多方法[1].这些方法中有很多虽然可以达到预期的效果,但是也会破坏原有聚合物的一些优良性能(如透明性、力学强度等).近些年来,通过端基功能化改变聚合物性能的研究越来越多,功能化端基位于链的两端,所以含量低,对聚合物本体性能影响小,而且还可以准确控制基团的数量[2].
在高分子聚合物中,聚醚类主要被用来生产聚氨酯(PU)泡沫塑料及表面活性剂,阴离子聚合是生产这一类聚合物常用的方法,进行功能化改性以后还可以用在生物医药等其他领域[3].聚酯类聚合物经常被用在制作生物降解材料、生物医学方面,各种功能化基团也被用来改性聚酯材料,其中尤以丙烯酸类和硫醇类居多,近年来利用反相碘转移聚合(RITP)制备功能化聚酯是大家研究的热点[4-5].在所有聚合物中,由于聚烯烃材料拥有价格低廉、易加工成型、综合性能优良等优点,成为一类产量最大,应用最广泛的高分子材料[6-7].然而,聚烯烃材料缺乏极性基团,具有一定的化学惰性,不利于印染、黏结以及与其他材料之间的相容.因此,长期以来,人们一直致力于通过一定的方法在聚烯烃分子中引入极性基团、反应性基团等对其进行功能化和高性能化处理.功能化以后的聚烯烃不仅性能得到很大改善,同时其应用范围也得到拓展[8-11].
近年来,链端功能化聚烯烃的合成方法主要有:(1)烯烃配位聚合过程链转移反应;(2)活性配位聚合;(3)活性阴离子聚合;(4)开环易位聚合中的链转移反应;(5)非烯烃聚合方法——叶立德同源聚合;(6)“Click”反应.其中,“Click”反应常与其他方法连用对聚合物的端基进行功能化的改性.
1 链转移反应
近年来,烯烃配位链转移反应在制备端基功能化聚烯烃领域被广泛应用,可以有效调节聚烯烃分子的结构和性能.现有的配位聚合链转移反应主要包括自发的β-H消除、β-Me消除,向聚合单体、外加链转移剂(CTA)转移,这些反应都可以用来制备端基功能化聚烯烃[12].
在催化乙烯聚合领域,链端功能化聚乙烯的生产通常受到效率、链端选择性转化等方面的限制,尤其是β-H的转移会使得链端失活.催化链增长反应(CCG)作为配位链转移聚合中的一种,在催化体系中可以非常便利地实现聚烯烃的链端功能化,而且CTA的链转移效率很高,这种方法最早由Ziegler[13]报道,工业上用于生产高级α-烯烃和高级醇.
Mazzolini等[14]利用丁烯辛基镁(BOMg),加入钕系催化剂催化乙烯聚合得到PE-Mg-PE,然后通过3种途径得到巯基封端的PE(图1[14]).(1)加入S8得到PE-Sx-PE,再通过LiAlH4还原得到巯基封端的聚乙烯.(2)加入碘得到PE-I聚合物,然后使用NaSH将末端的碘变为巯基.(3)PE-Mg-PE首先与二硫化物反应得到端基为硫酯基团的PE,然后使用LiAlH4将端基变为巯基.通过数据分析得出PE的相对分子质量越小巯基链端功能化率越高(Mn=1,300,g/mol,1,130,g/mol,链端功能化率70.6%,,72.7%,;Mn=2,270,g/mol,2,570,g/mol,链端功能化率56.9%,,63.4%,).

图1 合成PE-SH的3种不同路径Fig. 1 Three strategies to synthesize PE-SH
German等[15]则使用溴代十一碳烯(CH2=CH—(CH2)9—Br)合成了两端带有双键的二烷基镁((CH2=CH—(CH2)9)2—Mg),并以此作为链转移剂,加入Nd催化剂催化乙烯配位聚合得到(Vin-PE)2,Mg(Mn=1,150,g/mol,Mn,target=1,000,g/mol,PDI=1.13),进一步与碘或四乙基秋兰姆反应得到Vin-PE-I和Vin-PE-SC(S)NEt2,这两种聚合物的链端功能化率分别达到了96%,和87%,经过进一步的转化还可将链端功能性基团变为巯基、叠氮基和胺基.而且由于聚合物端基存在双键,还可以进行巯基-烯点击化学制备多种端基功能化聚乙烯[16-17].
Makio等[18]使用非共轭二烯和二乙基锌在Zr-FI催化下合成了特殊的单/多核烷基锌化合物.然后使用上述合成的多核烷基锌化合物,通过CCG反应,催化烯烃活性聚合,并且对聚合物中的Zn—C键进行功能化转换,得到了遥爪聚合物(图2[18]).
进一步拓展开来,硼氢化反应也是进行链端功能化的方法之一.烃基硼烷的硼原子具有空p轨道,显示较强的亲电性,可以和多种亲核试剂发生反应,其中最常用的就是硼烷与烯烃和炔烃的不饱和键加成反应.因此,在得到端羟基的聚烯烃以后,可以通过硼氢化反应进行链端基团的转换[19-20].目前最常用的硼氢化试剂主要有烷基硼、9-硼二环壬烷(9-BBN)、儿茶酚硼烷.2005年Hagiwara等[21]将双端含有丙烯基的聚丙烯(iPP-TVDs)分别与9-BBN和BH3·THF进行硼氢化反应,然后使用H2O2进行氧化断键得到HO-PP-OH,端基转化率最高分别达到了98%,和100%,之后又对羟基进行了一系列的改变,合成了无规共聚物(图3[21]).
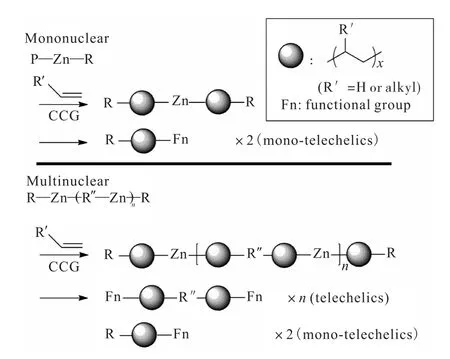
图2 利用乙烯在多核烷基锌化合物上催化链增长制备遥爪聚乙烯Fig. 2 Synthesis of telechelic olefin polymers via catalyzed chain growth on multinuclear alkylene zinc compounds
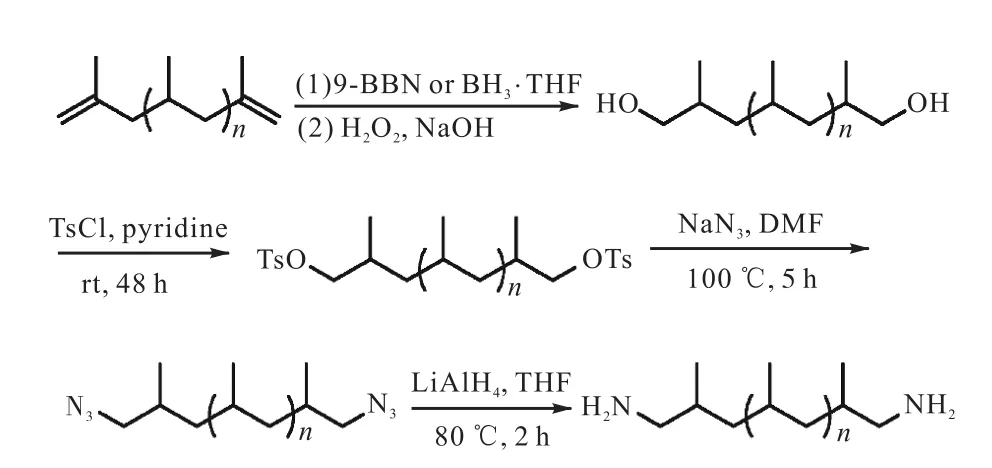
图3 硼氢化反应制备端基功能化聚丙烯Fig. 3 Synthesis of chain-end functionalized polypropylene via hydroboration
2 活性配位聚合
相对于其他的活性聚合方法,烯烃配位活性聚合不仅起步较晚,而且这种聚合对催化剂和反应条件的要求都较高,进一步制约了它的发展.但是近十几年来,随着茂/单茂金属催化剂、二元胺前过渡金属催化剂、后过渡金属钯催化剂、镍系催化剂、水杨醛亚胺前过渡金属催化剂(FI催化剂)的发展,使得这一领域发展势头猛进.
使用带有功能化基团的引发剂进行烯烃聚合是聚烯烃功能化的常用方法之一.Gottfried等[22]使用二亚胺金属钯催化剂催化乙烯聚合,在引发步骤就得到了链端功能化的聚乙烯,聚合完成后引入丙烯酸酯类单体插入活性中心,经三乙基硅烷裂解,得到远螯聚乙烯(图4[22]).使用相同的方法还合成出了链端功能化的聚丙烯(PDI<1.1)和聚己烯,在制备聚己烯时随着聚合时间的增加,相对分子质量分布增加(<100,min,PDI<1.08;3,h,PDI=1.15).为了改善聚乙烯的性能,他们还制备出了聚乙烯和聚十八烯的嵌段聚合物.
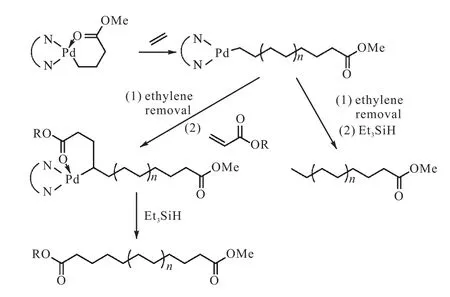
图4 丙烯酸酯单/双端基功能化聚烯烃的合成Fig. 4Synthesis of acrylate mono/difunctionalized polyolefins
Li等[23]也使用二亚胺金属钯催化剂催化乙烯活性聚合,聚合后加入苯乙烯衍生物插入活性中心,使得聚合物链从金属钯脱落,得到苯乙烯衍生物封端的远螯超支化聚乙烯(PDI=1.00~1.08),这些苯乙烯衍生物封端基团分别为苄基氯、4-甲基苯基、乙烯基苯.合成后的超支化聚乙烯可以被用作大分子引发剂或者大分子单体合成嵌段聚合物和其他结构更为复杂的聚合物.
Makio等[24]使用功能化引发剂,加入双苯氧基亚胺有机金属钛催化剂催化乙烯/丙烯聚合,得到一端含有羟基的聚乙烯和高间规聚丙烯(PDI=1.06).同时,聚合完成后再加入功能化终止剂,得到双端功能化的高间规聚丙烯(PDI≤1.08)(图5[24]).通过这种方法,只要使用不同的功能化引发剂和终止剂就可以得到带有各种功能化基团的聚烯烃.
得到一端为甲基,一端为其他功能性基团的聚烯烃以后,在甲基端进行功能基团转变的方法比较少.Schaffer等[25]以一端为酯基/羧基,一端为甲基的聚乙烯为起点,通过生物化学的方法,使用烷基单加氧酶,将甲基基团变为了羧基、醛基,进而得到双端功能化的聚乙烯.加入ω-转氨酶和醛去氢酶又可以将醛基分别转变为胺基和羧基.同时,也可以使用相同的方法,直接将PE进行端基功能化(图6[25]).
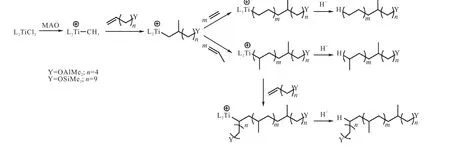
图5 双端功能化高间规聚丙烯的合成Fig. 5 Synthesis of difunctionalized high syndiotactic polypropylene
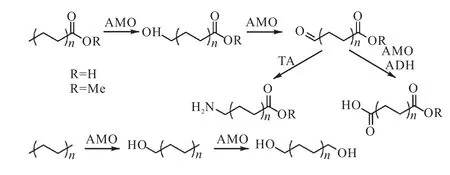
图6 利用生物酶进行端基转化反应制备功能化聚烯烃Fig. 6 Chemical modification of terminal group using biological enzyme
3 活性阴离子聚合
自从1956年Szware等[26-27]发现活性阴离子聚合以来,人们对非极性单体如苯乙烯、丁二烯等活性阴离子聚合进行了大量卓有成效的研究,建立了非极性单体的活性阴离子聚合法,并且进行了工业化生产.对于极性单体如甲基丙烯酸酯类,由于存在易于阴离子发生亲核加成的羰基,进行阴离子聚合时会导致引发剂失活,或发生环化反应形成混杂结构低聚物,难以实现活性聚合.但是经过研究发现,采用立体位阻很大的引发剂,或在聚合体系中添加无机盐类可以有效地抑制阴离子与羰基加成的副反应,从而实现活性聚合.近年来,多采用基团保护法,在实现阴离子聚合后将保护基团脱除也可顺利进行活性阴离子聚合.
Kimani等[28]合成了一系列不同相对分子质量的链端功能化聚丁二烯(主要为1,4-加成,92%,),分别使用具有低表面能的氟烷基和具有高表面能的羟基作为封端基团(图7[28]).其中,氟烷基封端的聚丁二烯直接通过阴离子活性聚合得到,而羟基封端的聚丁二烯则是结合了阴离子活性聚合和Click两种反应.核磁共振氢谱的结果显示链端功能化程度非常高(氟烷基封端的达到了99%,羟基封端的达到了97%,),同时接触角测量的数据显示高表面能的四羟基功能化聚丁二烯在硅片上的接触角比在聚四氟乙烯片上的小(与氟烷基功能化的聚丁二烯相反),这也刚好证明了功能化基团对聚合物表面润湿性的影响.

图7 活性阴离子聚合制备氟烷基封端的聚丁二烯Fig. 7Synthesis of fluorocarbon end-capped polybutadiene via living anionic polymerization
李勇等[29]用自制的叔丁基二甲基硅氧基丙基锂为引发剂,丁二烯为单体,加入四氢呋喃作为结构调节剂,在环己烷溶液中采用阴离子聚合法合成了线性端羟基聚丁二烯(L-HTPB),相对分子质量分布窄(PDI=1.06),平均官能度大于1.9.在此基础上,以甲基三氯硅烷为偶联剂合成了末端带有保护基团的星型聚丁二烯,盐酸酸化脱去保护基团生成羟基,得到星型端羟基聚丁二烯(S-HTPB).通过核磁表征,L-HTPB与S-HTPB微观结构含量基本相同,且含有较多的1,4-结构,证明从线性向星型聚丁二烯转化的过程中没有发生聚丁二烯微观结构的变化.
Zhang等[30]加入异丁基锂作为引发剂,进行丁二烯的阴离子聚合,进一步加入环氧乙烷得到相对分子质量分布窄的(PDI=1.04)羟基封端的聚丁二烯(PBd-t-OH).再利用端基转化反应得到PBd-t-Br和PBd-t-N3.他们又合成了两臂、三臂、四臂结构的3种交联剂(图8[30]),分别与PBd-t-N3进行“Click”反应得到3种星型聚合物,交联度分别为97%,、95.5%,和87%,而且相对分子质量越低,目标产物的转化率越高.
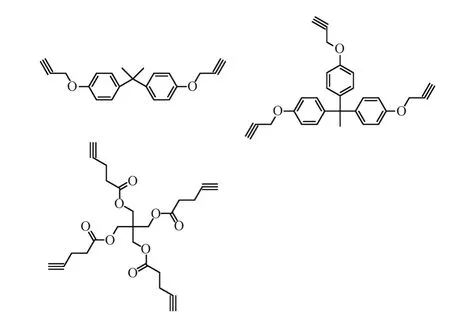
图8 两臂、三臂、四臂结构的交联剂Fig. 8 Linear(2-arm) and nonlinear(3-arm,4-arm) starlike crosslining agents
4 开环易位聚合
1967年Caldtron[31]首先提出了开环易位聚合(ROMP).近年来,由于ROMP具有活性聚合的特点,且反应条件温和、聚合速率高,因此受到了广泛的研究和应用.ROMP不是简单的链烯烃双键断裂的加成聚合,而是当环烯烃在催化剂作用下发生烯烃易位反应时,以头尾相接的方式连接成一个大分子,得到主链含双键的聚合物的一个过程[31].ROMP催化剂是以过渡金属为主催化剂,主族金属有机化合物为共催化剂组成的复合催化剂,主要有传统催化剂、水溶性催化剂和卡宾型催化剂3大类.
Pitet等[32]使用Ru卡宾催化剂引发顺式环辛烯(CEO)开环易位聚合,马来酸作为链转移剂(CTA),得到了羧基封端的聚环辛烯(PCEO),经过进一步的加氢反应得到了远螯聚乙烯(图9[32]).这种端羧基的远螯聚乙烯可以进一步进行链端功能化,并且与其他材料共混制备混合软材料.

图9 利用ROMP/氢化作用制备羧基封端的远螯聚环辛烯和聚乙烯Fig. 9Synthesis of carboxy-telechelic polycyclooctene and polyethylene using ROMP/hydrogenation
Diallo等[33]报道了Ru卡宾亚烷基催化剂引发3-烷基取代的环辛烯聚合,以三甲氧基硅单功能化烯烃为CTA,得到单/双端基功能化的聚合物.反应过程中聚烯烃结构多样化的形成主要经过两个阶段:首先,通过ROMP和RCM形成大分子或单功能化的大分子;然后,碳碳双键进行异构化得到双端基功能化大分子(图10[33]).CTA的性质、单体的取代基、催化剂的性质以及溶剂的选择都会影响反应的选择性和反应性.
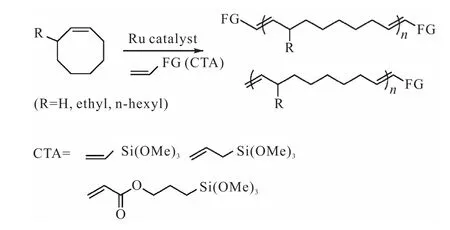
图10 α-和α,ω-三甲氧基硅遥爪聚烯烃的合成Fig. 10Synthesis of α-and α,ω-trimethoxysilane terminated polycyclooctene
Xiang等[34]使用Grubbs二代催化剂催化1,5-环二辛烯(COD)聚合,1,4-二乙酰氧基-2-丁烯为CTA,得到了双端乙酰氧基封端的聚环二辛烯(AcOPCOD-OAc),进一步加入甲醇钠,水解得到羟基封端的PCOD(HO-PCOD-OH).他们又以AcO-PCOD-OAc和HO-PCOD-OH为基础,通过传统的酶法开环聚合(eROP),加入十五酸内酯(PDL),一步法合成了嵌段共聚物AcO-LCL-OAc和HO-LCL-OH(图11[34]).
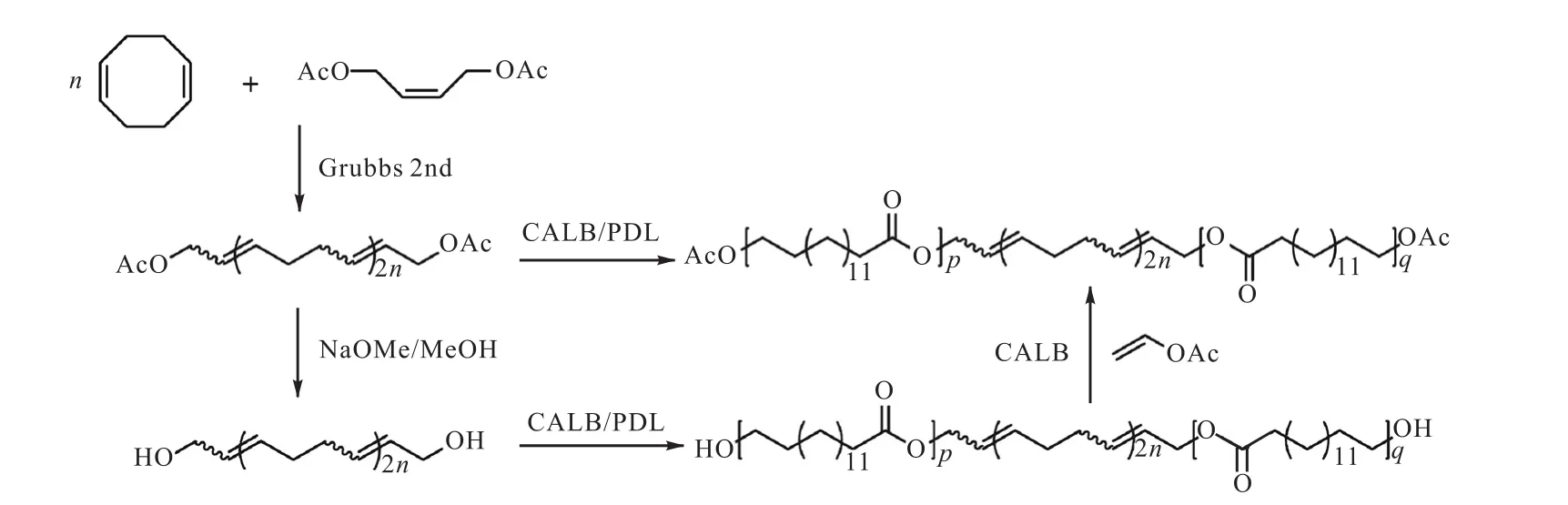
图11 双端基功能化聚烯烃的合成以及其在合成嵌段共聚物中的应用Fig. 11 Synthesis of difunctionalized polyolefins and block copolymer derived thereby
Martinez等[35]使用3-己烷-1-环辛烯和环辛烯两种单体进行开环易位聚合,马来酸作为CTA,合成了双端羧基功能化的嵌段共聚物,再催化加氢得到具有饱和主链的烯烃嵌段共聚物.借助之前合成的这种遥爪聚合物,又进行了扩链反应和交联反应,得到了聚烯烃高弹体,并且从动态力学和拉伸性能两方面对这种热固性高弹体的性能进行了研究.
5 叶立德同源聚合
叶立德同源聚合是由Shea等[36]于1997年首先提出的一种新型的合成聚烯烃碳链骨架的聚合方法.目前工业生产的一些聚合物都是利用碳碳双键聚合,这对于许多含极性基团的单体来说,单体的极性会使催化剂失活,其中一些虽然可以用自由基聚合来实现,但是却得不到立构规整的聚合物.但是叶立德同源聚合不同于传统的碳碳双键增长模式,它由路易斯酸性的硼烷和叶立德形成复合体系,以一次增长一个碳原子的方式构建成聚烯烃分子链,如图12[37]所示.这种方法不仅具有活性聚合的特征,而且没有明显的链转移和链终止反应,能够很好地控制聚合物的相对分子质量及其分布.同时,如果使用带有功能性基团的烷基硼,则可以直接合成双端功能化的遥爪聚乙烯.
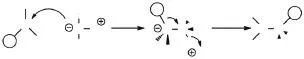
图12 叶立德同源聚合机理Fig. 12 Mechanism of the polyhomologation
Busch等[38]、Luo 等[39]利用硼烷引发/催化的硫叶立德活性聚合合成了远螯聚亚甲基聚合物.通过α-烯烃的硼氢化反应得到功能化的星型有机硼聚合物,再经过二水氧化三甲胺(TAO)氧化,得到相对分子质量范围在1,000~17,000,相对分子质量分布很窄(PDI<1.1)的主链链端功能化的远螯聚亚甲基聚合物.
在叶立德同源聚合中,可以直接通过使用带有功能性基团的硼烷获得功能化聚烯烃,因此,对于硼烷的开发也是十分重要的.Zhang等[40]利用乙烯基与硼烷的硼氢化反应得到三乙基硼,然后以它作为引发剂和催化剂,引发硫叶立德活性聚合形成一系列功能化的星型有机硼聚合物,再经过氧化断键,得到α,ω-端基功能化的聚乙烯.利用相同的聚合过程,还得到了PEO-b-PM双嵌段共聚物以及PM-b-PDMS-b-PM三嵌段共聚物.PM-OH与2-溴-2-甲基丙酰溴反应得到PM-Br,PM-Br可以作为引发剂引发PS进行原子转移自由基聚合(ATRP)得到PM-PS嵌段共聚物.
Alkayal等[41]首先用9-蒽基甲基烯丙基醚与硼烷反应得到带有功能性基团的三臂结构的硼烷,再经过硫叶立德同源聚合,合成了一端为蒽基,一端为羟基的聚亚甲基聚合物(α-anthracene-ω-hydroxy-polymethylene),然后以它为底物通过呋喃保护的马来酰亚胺与蒽的Aiels-Alder反应合成了几种双/三嵌段聚合物.这两种方法的结合为以聚乙烯为底物的复杂大分子结构材料的合成开拓了新视野.
赵亮等[42]通过二乙烯基苯的硼氢化反应,制备了含有苯乙烯功能基团的三烷基硼中间体,并以此为引发剂进行叶立德活性聚合,最终得到基于聚亚甲基的一端含有羟基另一端含有苯乙烯基的大分子单体(St-PM-OH),高温GPC的检测结果显示Mn=1,385,g/mol,PDI=1.3,相对分子质量的可控性好.
Zhang等[43]用甲基苯基二乙烯基硅烷与叔己基硼烷反应合成了一种新的含硅的环状硼烷,这种类型的硼烷同时拥有两个引发位点,可以制备以PE为主链的复杂大分子结构聚合物.使用这种硼烷与硫叶立德进行反应,再经过TAO氧化断键,可以得到双端羟基的远螯聚乙烯(Mn,NMR=1,400,g/mol,PDI=1.25)(图13[43]).合成具有两个这种环状的硼烷化合物,经过叶立德聚合则可以获得四臂的端羟基星型聚乙烯.另外,他们结合ATRP和叶立德同源聚合两种方法,还得到了两臂为PE,一臂为PS的星型聚合物以及以PE为支链的梳型聚合物.
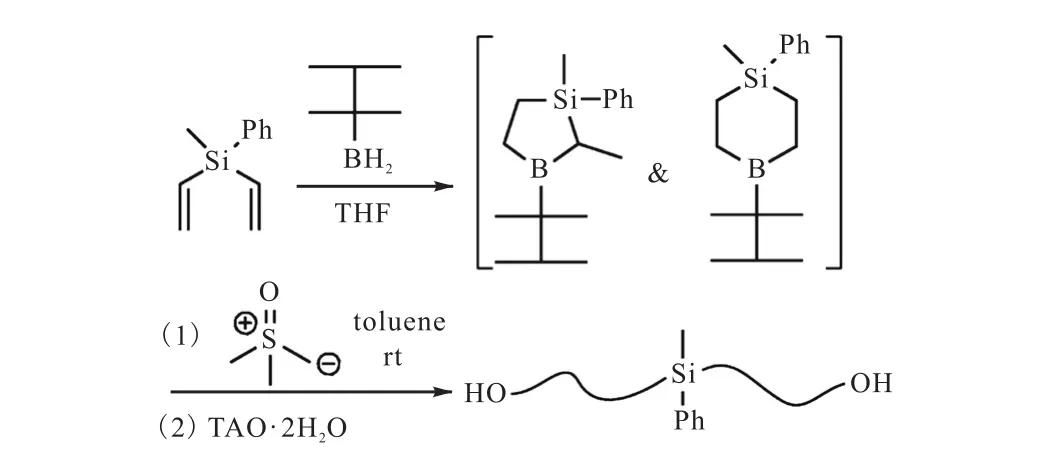
图13 环状硼烷与叶立德反应制备双端羟基功能化聚乙烯Fig. 13 Synthesis of difunctionalized polyethylene via B-thexyl-silaboracyclic structure in polyhomologation
近十几年来,聚合物分子刷的研究受到了广泛的关注.所谓聚合物分子刷是指高密度地将聚合物分子链的一端连接于各种界面或聚合物链上形成的一种特殊均聚或共聚高分子体系.这类聚合物可以很好地控制和改变界面或表面的物性,通过改变聚合物分子刷的结构或组成可以控制聚合物刷的聚集形态及形态转换,因而广泛应用在分子自组装、药物输送、有机金属纳米材料等领域.
Zhang等[44-45]将叶立德同源聚合与ROMP两种方法结合在一起合成了几种以PE 为主链的无规、嵌段、双层聚合物分子刷.采用三乙基硼为引发剂进行叶立德聚合得到PE-OH,PE-OH与5-降冰片烯-2-甲酸进行酯化反应,将羟基变为降冰片烯基,然后以此作为大分子单体,在Grubbs催化剂作用下进行开环易位聚合得到聚合物分子刷.
除了硫叶立德外,目前研究比较多的还有胂叶立德和磷叶立德.Mondière等[46]合成了以下4种胂叶立德,并且使用它们首先进攻三乙基硼烷形成两性离子化合物,然后脱去三苯基胂,通过分子内的烷基迁移形成具有烯丙基的有机硼化合物,重复此过程进行胂叶立德同源聚合,最后加入H2O2氧化断键,得到具有端羟基的poly(2-substituted-1-propenylene)(图14[46]).利用相同的聚合过程,还可以加入不同的胂叶立德得到嵌段聚合物.
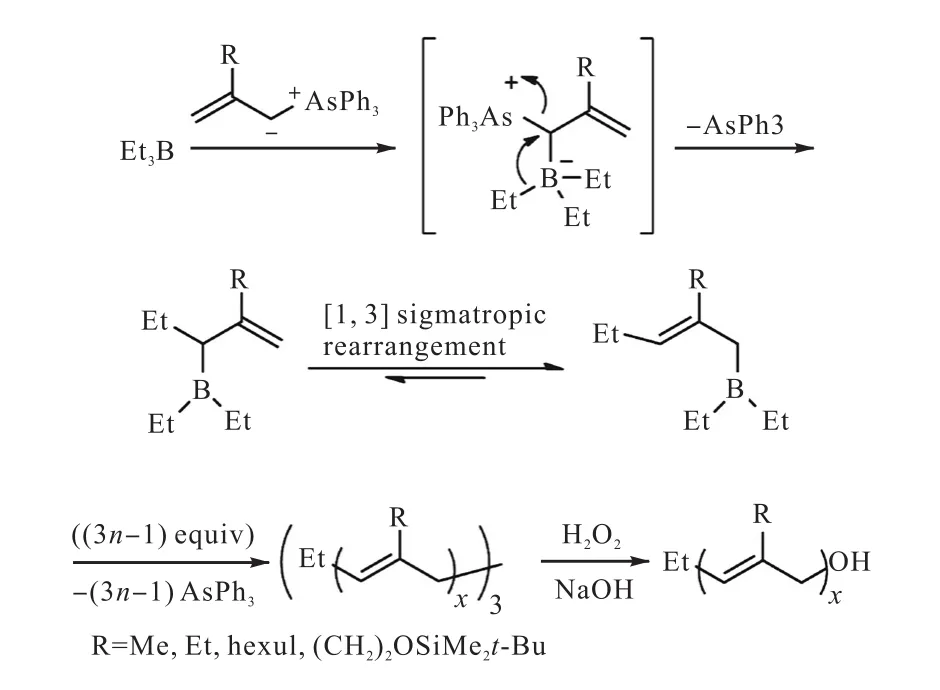
图14 三乙基硼与胂叶立德反应合成链端含有羟基的poly(2-substituted-1-propenylene)Fig. 14Synthesis of poly(2-substituted-1-propenylene)s from Allylic Arsonium Ylide
6 “Click”反应
2001年美国诺贝尔化学奖获得者Sharpless发展出一种名为“click chemistry”的新技术,其所具有的高效和高控制性,在化学合成领域掀起了一场风暴,目前它已经成为药物开发和生物医用材料等诸多领域最吸引人的发展方向.点击化学反应主要有4种类型:(1)环加成反应;(2)亲核开环反应;(3)碳碳多键的加成反应;(4)非醇醛的羰基化反应.其中,尤以第一种类型中的铜催化叠氮-炔基环加成反应(CuAAC)最为典型,应用也最广泛.
Magenau等[47]通过硫醇-烯点击化学合成了一系列单/双官能化的聚异丁烯,其中功能化基团包括卤素原子、羟基、羧基、胺基.反应需要的时间比较短(<10,min),不需要复杂的纯化过程和苛刻的反应条件,并且降低反应温度(冰浴)还会促进功能化基团的转换(≥97%,)(图15[47]).
Huang等[48]通过活性配位聚合首先合成了苯乙烯封端的聚丙烯(iPP-t-St),iPP-t-St与氯化氢加成得到卤素封端的聚丙烯(iPP-t-Cl),可以用来引发MMA进行ATRP聚合得到嵌段共聚物(iPP-b-PMMA)[49].将卤素端基变为叠氮基后,与端炔基进行CuAAc反应,可以获得星型等规聚丙烯.
图15 通过硫醇-烯点击化学进行端基转化反应制备功能化聚异丁烯Fig. 15 Polyisobutylene end functionalization using thiolene click chemistry
点击化学主要是通过小单元的拼接,快速可靠完成各类分子的化学合成,大分子之间的点击化学理论上可行,但主要困难在于产物的分离.Malke等[50]首先通过活性碳正离子聚合(LCCP)得到了两种不同的α,ω-链端功能化的聚异丁烯(PIB),一种链端为炔基(Mn=4,700,g/mol,PDI=1.39),另一种链端为伯胺基(Mn=5,000,g/mol,PDI=1.30),然后通过酰胺化反应,加入β转角二肽(BTD)与链端含有胺基的PIB进行端基转化反应,得到聚合物PIB-BTD.由于PIBBTD中链端含有叠氮基团,故可以与链端炔基的PIB进行两个大分子之间的Click反应(图16[50]).
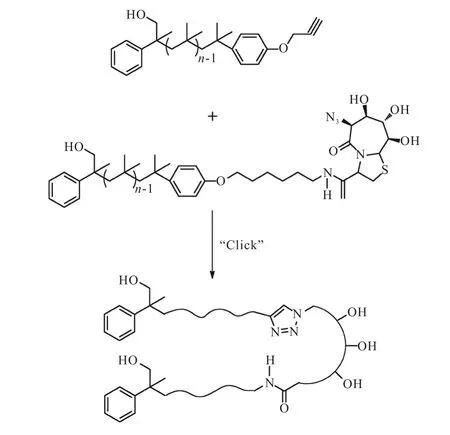
图16 聚异丁烯大分子之间的“Click”反应Fig. 16 Cilck chemistry between polyisobutylene macromolecules
在酰胺化和Click两个反应过程中,BTD和PIB-BTD都受到空间构象的限制,使得羧基和叠氮基两个反应位点周围的空间变小,从而降低了它们的反应活性,因此在反应中加入DIPEA以减小空间位阻. 此外,由于PMDETA对叠氮基有特殊的空间屏蔽效应,在反应中加入以后,没有目标产物生成,这一点也刚好证明了空间因素会对这一反应的显著影响.他们还使用SEC和1H NMR对反应物和产物进行了测定,证明了Click反应的成功和产物的纯度.
7 总结与展望
烯烃配位聚合过程链转移反应需要链转移剂,且每种链转移剂都有一定的局限性,存在功能化效率不高、相对分子质量分布宽等缺陷;对于活性配位聚合,虽然可以很好地调控聚合物的结构,但受到所用催化剂发展的限制使得这种方法的发展也受限,而且一般的金属催化剂比较昂贵.阴离子活性聚合对功能化聚烯烃结构的可控性强,但是反应条件比较苛刻,很多功能性基团难以与聚合使用的引发剂相容.开环易位聚合反应条件比较温和,反应速率快,但是只适用于环的张力较大的单体.相比较而言,叶立德同源聚合可以很好地控制聚合物相对分子质量,而且得到的聚合物相对分子质量分布较窄,端基功能化率高,反应条件温和,不需要外加催化剂,拥有广阔的发展前景.在今后的研究中,可以更多地与Click反应、硼氢化反应以及现有的一些生物化学等方法相结合,开发更加简洁高效、绿色环保的链端功能化聚烯烃合成新路径,提高聚烯烃材料在各方面的现实应用.
[1] 王国全,王秀芬. 聚合物改性[M]. 2版. 北京:中国轻工业出版社,2008.
[2] Koberstein J T. Molecular design of functional polymer surfaces[J]. Journal of Polymer Science Part B:Polymer Physics,2004,42(16):2942-2956.
[3] Wurm F,Klos J,Räder H J,et al. Synthesis and noncovalent protein conjugation of linear-hyperbranched PEG-poly(glycerol)α,ωn-telechelics[J]. Journal of the American Chemical Society,2009,131(23):7954-7955.
[4] Bouilhac C,Chirat M,Joly-Duhamel C,et al. Reverse iodine transfer polymerization(RITP)of 1,1,2,2-tetrahydroperfluorodecyl acrylate in supercritical carbon dioxide[J]. Macromolecular Chemistry and Physics,2013,214(20):2259-2265.
[5] David G,Boyer C,Tonnar J,et al. Use of iodocompounds in radical polymerization[J]. Chemical Reviews,2006,106(9):3936-3962.
[6] Vasile C. Handbook of Polyolefins[M]. 2nd ed. New York:Marcel Dekker,2000:249.
[7] 洪定一. 塑料工业手册:聚烯烃[M]. 北京:化学工业出版社,1999:296
[8] Chung T C. Synthesis of functional polyolefin copolymers with graft and block structures[J]. Progress in Polymer Science,2002,27(1):39-85.
[9] 陈健壮,崔崑,张淑媛,等. 主链链端功能化聚烯烃的设计、合成及其应用[J]. 化学进展,2008,20(11):1740-1750.
[10] Povie G,Tran A T,Bonnaffé D,et al. Repairing the thiolene coupling reaction[J]. Angewandte Chemie International Edition,2014,53(15):3894-3898.
[11] Franssen N M G,Reek J N H,de Bruin B. Synthesis of functional ‘polyolefins’:State of the art and remaining challenges[J]. Chemical Society Reviews,2013,42(13):5809-5832.
[12] 张勇杰,李化毅,董金勇,等. 端基功能化聚烯烃的合成与应用[J]. 化学进展,2014,26(1):110-124.
[13] Ziegler K. Aluminium-organische synthese im bereich olefinischer kohlenwasserstoffe[J]. Angewandte Chemie,1952,64(12):323-329.
[14] Mazzolini J,Mokthari I,Briquel R,et al. Thiol-endfunctionalized polyethylenes[J]. Macromolecules,2010,43(18):7495-7503.
[15] German I,Kelhifi W,Norsic S,et al. Telechelic polyethylene from catalyzed chain-growth polymerization[J]. Angewandte Chemie International Edition,2013,52(12):3438-3441.
[16] Mazzolini J,Boyron O,Monteil V,et al. Polyethylene end functionalization using radical-mediated thiol-ene chemistry:Use of polyethylenes containing alkene end functionality[J]. Macromolecules,2011,44(9):3381-3387.
[17] Norsic S,Thomas C,D'Agosto F,et al. Divinyl-endfunctionalized polyethylenes:Ready access to a range of telechelic polyethylenes through thiol-ene reactions[J]. Angewandte Chemie International Edition,2015,54(15):4631-4635.
[18] Makio H,Ochiai T,Mohri J,et al. Synthesis of telechelic olefin polymers via catalyzed chain growth on multinuclear alkylene zinc compounds[J]. Journal of the American Chemical Society,2013,135(22):8177-8180.
[19] Brown H C,Chandrasekharan J. Hydroboration. 65. Relative reactivities of representative alkenes and alkynes toward hydroboration by catecholborane[J]. The Journal of Organic Chemistry,1983,48(25):5080-5082.
[20] Garrett C E,Fu G C. Hydroboration of olefins with catecholborane at room temperature in the presence of N,N-dimethylacetamide[J]. Journal of Organic Chemistry,1996,61(10):3224-3225.
[21] Hagiwara T,Saitoh H,Tobe A,et al. Functionalization and applications of telechelic oligopropylenes:Preparation of α,ω-dihydroxy-and diaminooligopropylenes[J]. Macromolecules,2005,38(25):10373-10378.
[22] Gottfried A C,Brookhart M. Living and block copolymerization of ethylene and α-olefins using palladium(Ⅱ)-α-diimine catalysts[J]. Macromolecules,2003,36(9):3085-3100.
[23] Li S Y,Ye Z B. Synthesis of narrowly distributed ωtelechelic hyperbranched polyethylenes by efficient endcapping of Pd-diimine-catalyzed ethylene “living” polymerization with styrene derivatives[J]. Macromolecular Chemistry and Physics,2010,211(17):1917-1924.
[24] Makio H,Fujita T. Synthesis of chain-end functionalized polyolefins with a bis(phenoxy imine)titanium catalyst[J]. Macromolecular Rapid Communications,2007,28(6):698-703.
[25] Schaffer S,Haas T. Biocatalytic and fermentative production of α,ω-bifunctional polymer precursors[J]. Organic Process Research & Development,2014,18(6):752-766.
[26] Szware M. ‘Living’ polymers[J]. Nature,1956,178:1168-1169
[27] Szwarc M,Levy M,Milkovich R. Polymerization initiated by electron transfer to monomer. A new method of formation of block polymers[J]. Journal of the American Chemical Society,1956,78(11):2656-2657.
[28] Kimani S M,Hardman S J,Hutchings L R,et al. Synthesis and surface activity of high and low surface energy multi-end functional polybutadiene additives[J]. Soft Matter,2012,8(12):3487-3496.
[29] 李勇,易建军,陈继明,等. 线性/三臂星型端羟基聚丁二烯的活性负离子聚合及其表征[J]. 高分子学报,2015(5):508-514.
[30] Zhang G,Zheng J,Liu F,et al. Synthesis of star-like polybutadienes by a combination of living anionic polymerization and “click” coupling method[J]. Chinese Journal of Polymer Science,2014,32(6):731-742.
[31] 张留成,闫卫东,王家喜. 高分子材料进展[M]. 北京:化学工业出版社,2005:6.
[32] Pitet L M,Hillmyer M A. Carboxy-telechelic polyolefins by ROMP using maleic acid as a chain transfer agent[J]. Macromolecules,2011,44(7):2378-2381.
[33] Diallo A K,Michel X,Fouquay S,et al. α-Trialkoxysilyl functionalized polycyclooctenes synthesized by chaintransfer ring-opening metathesis polymerization[J]. Macromolecules,2015,48(20):7453-7465.
[34] Xiang S D,Zhang Q P,Zhang G,et al. Facile synthesis of block copolymers by tandem ROMP and eROP from esters precursors[J]. Biomacromolecules,2014,15(8):3112-3118.
[35] Martinez H,Hillmyer M A. Carboxy-telechelic polyolefins in cross-linked elastomers[J]. Macromolecules,2014,47(2):479-485.
[36] Shea K J,Walker J W,Zhu H,et al. Polyhomologation. A living polymethylene synthesis[J]. Journal of the American Chemical Society,1997,119(38):9049-9050.
[37] Shea K J. Polyhomologation:The living polymerization of ylides[J]. Chemistry:A European Journal,2000,6(7):1113-1119.
[38] Busch B B,Staiger C L,Stoddard J M,et al. Living polymerization of sulfur ylides. Synthesis of terminally functionalized and telechelic polymethylene[J]. Macromolecules,2002,35(22):8330-8337.
[39] Luo J,Shea K J. Polyhomologation. A living C1 polymerization[J]. Accounts of Chemical Research,2010,43(11):1420-1433.
[40] Zhang H F,Alkayal N,Gnanou Y,et al. Polymethylenebased copolymers by polyhomologation or by its combination with controlled/living and living polymerizations[J]. Macromolecular Rapid Communications,2014,35(4):378-390.
[41] Alkayal N,Hadjichristidis N. Well-defined polymethylene-based block co/terpolymers by combining anthrax cene/maleimide diels-alder reaction with polyhomologation[J]. Polymer Chemistry,2015,6(27):4921-4926.
[42] 赵亮,陈健壮,史丽萍,等. 叶立德活性聚合制备基于聚亚甲基的大分子单体[J]. 化学学报,2010,69(5):591-595.
[43] Zhang Z,Zhang H F,Gnanou Y,et al. Polyhomologation based on in situ generated boron-thexyl-silaboracyclic initiating sites:A novel strategy towards the synthesis of polyethylene-based complex architectures[J]. Chemical Communications,2015,51(49):9936-9938.
[44] Zhang H F,Gnanou Y,Hadjichristidis N. Well-defined polyethylene molecular brushes by polyhomologation and ring opening metathesis polymerization[J]. Polymer Chemistry,2014,5(22):6431-6434.
[45] Zhang H F,Zhang Z,Gnanou Y,et al. Well-defined polyethylene-based random,block,and bilayered molecular cobrushes[J]. Macromolecules,2015,48(11):3556-3562.
[46] Mondière R,Goddard J P,Carrot G,et al. Synthesis of poly(2-substituted-1-propenylene)s from allylic arsonium ylides[J]. Macromolecules,2005,38(3):663-668.
[47] Magenau A J D,Chan J W,Hoyle C E,et al. Facile polyisobutylene functionalization via thiol-ene click chemistry[J]. Polymer Chemistry,2010,1(6):831-833.
[48] Huang H H,Niu H,Dong J Y. Synthesis of star isotactic polypropylene using click chemistry[J]. Macromolecules,2010,43(20):8331-8335.
[49] Huang H H,Niu H,Dong J Y. Synthesis of i-PP-based functional block copolymer by a facile combination of styryl-capped i-PP and ATRP[J]. Journal of Polymer Science Part A:Polymer Chemistry,2010,48(24):5783-5789.
[50] Malke M,Barqawi H,Binder W H. Synthesis of an amphiphilic β-turn mimetic polymer conjugate[J]. ACS Macro Letters,2014,3(4):393-397.
责任编辑:周建军
Design and Synthesis of α,ω-Chain End Functionalized Polyolefins
JIANG Tao1,XU Fang1,2,DONG Peng1,2,MA Zhi2
(1.College of Chemical Engineering and Materials Science,Tianjin University of Science & Technology,Tianjin 300457,China;2.Shanghai Institute of Organic Chemistry,Chinese Academy of Sciences,Shanghai 200032,China)
Design and synthesis of α,ω-chain end functionalized polyolefins have become one of the hot research topic in recent years.There are some methods for synthesizing α,ω-chain end functionalized polyolefins.The living polymerization generally requires appropriate metal catalyst,the product has narrow molecular weight distribution,and the polymer chain is still active after the monomer consumption.Polymerization can still go on when adding new monomer.It can be usedto prepare block copolymer.Moreover,difunctionalized polyolefins can be got by using terminator.By introuducing trialkylborane in polyhomologation,reactive group terminated polyolefins can be obtained.Successive chemical modification of the chain end group can result in various polyolefins.In addition,chain transfer reaction in the polyolefin polymerization,ringopening metathesis polymerization,as well as the combination of both methods with “Click” reaction are all the main ways of synthesizing chain end functionalized polyolefins.The polyolefins not only can be used as polymer modifier,but also are starting points for other more complex structures,such as block copolymers,graft copolymers,and more complex polymer brushes.
polyolefins;α,ω-chain end functionalized;chain transfer reaction;living polymerization;polyhomologation;“Click” reaction
O632.1
A
1672-6510(2016)03-0001-10
10.13364/j.issn.1672-6510.20150230
2015-12-05;
2016-03-31
国家自然科学基金资助项目(21374130)
姜 涛(1972—),男,黑龙江人,教授,jiangtao@tust.edu.cn.
