HPLC法测定大鼠血浆及粪便中新疆紫草3种萘醌类色素的含量
2016-07-27古力扎努阿合买提买尔旦马合木提
古力扎努·阿合买提, 买尔旦·马合木提
(新疆医科大学1药学院; 2附属肿瘤医院, 乌鲁木齐 830011)
HPLC法测定大鼠血浆及粪便中新疆紫草3种萘醌类色素的含量
古力扎努·阿合买提1, 买尔旦·马合木提2
(新疆医科大学1药学院;2附属肿瘤医院, 乌鲁木齐830011)
摘要:目的建立测定大鼠血浆及粪便中新疆紫草3种萘醌类成分含量的高效液相色谱法(HPLC法)。方法采用COSMOIL C18-AR-Ⅱ(4.6 mm×250 mm,5 μm)色谱柱,流动相A乙腈(A)-0.15%甲酸(B)(v/v)溶液,进行洗脱,流速1.0 mL/min,检测波长275 nm ,柱温25℃,检测时间35 min。结果血浆中左旋紫草素在0.40~6.40 μg、乙酰紫草素在25.0~600.0 μg、β,β′-二甲基丙烯酰紫草素在25.0~600.0 μg 范围内均与峰面积呈线性关系,左旋紫草素回收率为97.78%~100.1%、乙酰紫草素为100.1%~100.2%、β-β′-二甲基丙烯酰紫草素为97.78%~100.1%,日间、日内RSD均<1.5%。粪便中各组分在10~400 μg/mL范围内与峰面积呈线性关系,平均回收率分别为(78.2±2.5)%、(82.0±2.9)%和(79.5±3.5)%。0~12、12~24、24~48 h粪便中左旋紫草素绝对回收量分别为23.61、38.28、4.13 mg;乙酰紫草素分别为7.20、70.78、11.53 mg;β-β′二甲基丙烯酰紫草素分别为46.32、60.15、16.88 mg。结论HPLC法准确可靠,重复性好,适用于测定血浆及粪便中新疆紫草萘醌类成分含量。
关键词:新疆紫草; 左旋紫草素; 乙酰紫草素; β,β′-二甲基丙烯酰紫草素; HPLC; 大鼠血浆; 大鼠粪便
新疆紫草[Arnebia euchroma (Royle) Johnst]习称软紫草,又名假紫草、新疆软紫草、新疆假紫草, 为紫草科多年生草本植物新疆紫草的根, 分布于我国新疆、甘肃及西藏西部地区, 是药材紫草的主要来源[1]。其具有凉血、活血、解毒透疹的功效,被临床广泛应用[2]。新疆紫草中含有多种化学成分,包括萘醌类、苯醌类、生物碱类、苯酚、酚酸类、三萜酸、甾醇类、黄酮类以及多糖类等物质。目前研究较多的是脂溶性很强的萘醌类化合物。萘醌类化合物主要有去氧紫草素(deoxyshikonin)、β,β-甲基丙烯酰紫草素(β,β-dimethylacrylshikonin,DS)、β-羟基异戊酰紫草素(β-hydroxyisovalerylshikonin)、乙酰紫草素(acetylshikonin,AS)、左旋紫草素(shikonin,SK)、去氢阿卡宁(dehydroalkanin)和β-羟基异戊酰阿卡宁(β- hydroxyisovalerylalkanin)等物质[3],其具有抗菌抗病毒、抗炎、镇痛、免疫调节、抗肿瘤、保肝、止血等作用[4-6]。本研究旨在建立HPLC法测定大鼠生物样品血浆及粪便中新疆紫草3种萘醌类成分含量,现报道如下。
1仪器与试药
1.1实验仪器LC-2010C高效液相色谱仪(日本岛津),CLASS-VP V6.14 SP1数据工作站(日本岛津),BP211D型十万分之一电子天平(德国Sartorius公司),SK3300H型超声波清洗仪(上海科导超声仪器有限公司),DT-500A型电子天平(常熟市金羊砝码仪器有限公司),TDL-4型台式离心机(上海安亭科学仪器厂),EDTA k2抗凝真空采血管(奥地利VACUETTE)。
1.2试药新疆紫草对照药材(中国食品药品检定研究院,批号:121430-201103),左旋紫草素对照品(中国药品生物制品检定所,批号:110769-200405),乙酰紫草素对照品(德瑞试剂公司,批号:20130507-2),β,β′-二甲基丙烯酰紫草素对照品(中国药品生物制品检定所,批号:111689-200503)。新疆紫草药材(购自新疆参茸中药饮片公司批号:15072402),经新疆医科大学药学院天然药物化学/生药学教研室帕丽达·阿布力孜教授鉴定为拟紫草属植物新疆紫草(Arnebia euchroma (Royle) Johnst)的根。乙腈为色谱纯(美国Fisher公司),水为超纯水,其余试剂均为分析纯。
1.3实验动物SD大鼠由新疆医科大学实验动物中心提供,许可证号SCXK(新)2011-0004,SPF级,体质量180~220 g。
2方法与结果
2.1对照品溶液的制备分别精密称取左旋紫草素、乙酰紫草素、β,β′-二甲基丙烯酰紫草素对照品适量,分别溶于甲醇中,制成100 mg/mL的对照品储备液。再分别量取储备液适量,用甲醇稀释制成含左旋紫草素、乙酰紫草素和β.β′-二甲基丙烯酰紫草素分别为1.00、60.0 和60.0 mg/mL的混合对照品溶液,备用。取本品适量,按递减法,分别用流动相稀释制成含左旋紫草素0.01、0.02、0.04、0.08、0.16、0.32、0.64 mg/mL,乙酰紫草素0.125、2.50、5.00、10.0、20.0、40.0、60.0 mg/mL,β,β′-二甲基丙烯酰紫草素0.125、2.50、5.00、10.0、20.0、40.0、60.0 mg/mL的混合对照品系列溶液,-20℃保存备用。
2.2供试品溶液的制备分别称取对照药材和供试品药材粉末各5.0 g,各加入12倍量(V/V)的95%乙醇溶液,置于150 mL具塞锥形瓶中,混匀,室温放置,密封遮光,冷浸8 h后,过滤并收集滤液,旋转蒸发仪减压浓缩至约5.0 mL,用5.0 mL 95%乙醇润洗,合并浓缩液和润洗液,置于真空干燥箱40℃干燥12 h,得干浸膏。分别称取干浸膏粉末0.1 g,加入1.0 mL甲醇,称重,超声使其完全溶解,冷却至室温,补足重量,过(0.45 μm)微孔滤膜,作为对照药材和样品的供试品溶液,备用。
2.3色谱条件COSMOILC18-AR-Ⅱ色谱柱(4.6 mm×250 mm,5 μm),流动相乙腈(A)-0.15%甲酸(B)(v/v)溶液,洗脱流程0~10 min,A(%)=60%;10~15 min,A(%)=60%~70%;15~30 min,A(%)=70%;30~35 min,A(%)=70%~60%,流速1.0 mL/min;柱温25℃,检测波长275 nm ,检测时间35 min,进样量20 μL。
2.4血浆样品的处理取SD大鼠血浆48 μL于离心管中,加入对照品药材溶液2.0 μL,涡旋混合1 min,加入等体积的稀盐酸酸化,10 000 r/min离心2 min,取上清液50 μL于另一离心管中,加入乙腈50 μL涡旋混合1 min,10 000 r/min离心,取上清液,氮气吹干,加入50 μL乙腈复溶,10 000 r/min离心10 min,取上清液备用。
2.4.1专属性考察精密量取1 mg/mL的对照药材储备液0.2 mL于离心管中,挥干溶剂,加入空白血浆50 μL混匀。另取空白血浆、对照药材和供试品溶液各50 μL与不同的离心管中,按“2.4”项下方法处理,按“2.3”项下色谱条件检测,记录色谱峰。在该色谱条件下,左旋紫草素、乙酰紫草素、β,β′-二甲基丙烯酰紫草素分离效果好,空白血浆中的内源性物质不干扰测定,结果见图1~4。


1:左旋紫草素;2:乙酰紫草素;3:β,β-二甲基丙烯酰紫草素

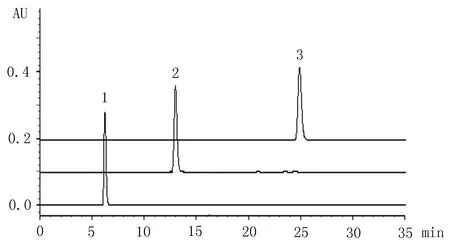
1:左旋紫草素;2:乙酰紫草素;3:β,β-二甲基丙烯酰紫草素
2.4.2标准曲线与最低检测限取空白血浆 7份,每份50 μL,分别加入混合对照品系列溶液10 μL,使浓度分别成为:左旋紫草素0.10、0.20、0.40、0.80、1.60、3.20、6.40 mg/mL,乙酰紫草素和β,β′-二甲基丙烯酰紫草素成为:1.25、25.0、50.0、100.0、200.0、400.0、600.0 μg/mL,按“2.4”项下方法处理,按“2.3”项下的色谱条件检测,记录色谱峰,以归一化法计算处理。以各组分相应峰面积为纵坐标(Y),各组分加入量为横坐标(X),进行线性回归,左旋紫草素在0.40~6.40 mg/mL范围内与峰面积呈线性关系,线性方程为:Y=154.75X+896.75,R=0.997 0,最低检测限为0.40 mg/mL;乙酰紫草素在25.0~600.0 μg范围内与峰面积呈线性关系,线性方程为:Y=378.09 X+9 611.3,R=0.994 3;最低检测限为25 μg/mL;β,β′-二甲基丙烯酰紫草素在25.0~600.0 μg范围内与峰面积呈线性关系,线性方程为:Y=1 005.9X-1 832,R=0.992 1,最低检测限为25 μg/mL。
2.4.3精密度试验取空白血浆,按“2.4”项下方法分别制备左旋紫草素、乙酰紫草素和β,β′-二甲基丙烯酰紫草素低、中、高3个浓度(含左旋紫草素0.10、0.80、6.40 μg/mL;含乙酰紫草素和β,β′-二甲基丙烯酰紫草素均为1.25、100.00、600.00 μg/mL)溶液各6份,每个浓度的样品分别每天测定1次,连续测定3 d,以当日标准曲线计算测定样品的浓度,分别计算日内精密度与日间精密度,见表1。
2.4.4回收率试验取“2.4.3”项下低、中、高3个浓度的样品,以空白血浆加混合标准品溶液提取处理样品与直接测定的对照品的峰面积比值计算提取回收率,左旋紫草素低、中、高3个样品的回收率分别为(97.78±2.50)%、(99.20±2.50)%和(100.10±2.51)%;乙酰紫草素为(100.10±2.12)%、(100.00±3.12)%和(100.2±3.01)%;β,β′-二甲基丙烯酰紫草素为(97.78±2.50)%、(99.90±2.12)%和(100.10±3.20)%。
2.4.5稳定性试验取精“2.4.3”项下低、中、高3个浓度的血浆样品,室温放置6 h,反复冻融3次,-20℃冰冻保存20 d,按“2.3”项下方法处理后进样,结果,本实验条件下,血浆样品中左旋紫草素、乙酰紫草素和β,β′-二甲基丙烯酰紫草素反复冻融3次仍保持稳定,见表2。
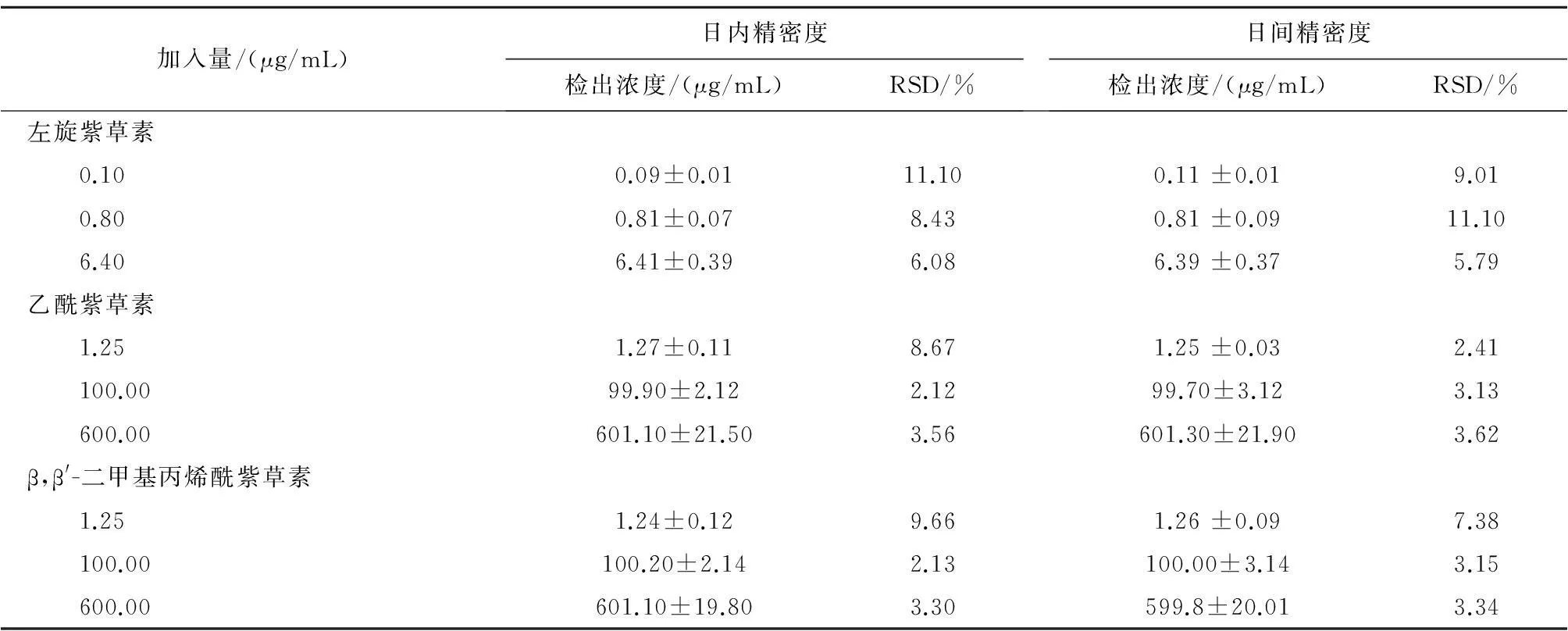
表1 精密度试验

表2 稳定性试验结果
2.5粪便样品的处理
2.5.1粪便样品的处理称取空白大鼠粪便30 mg于离心管中,加入乙酸乙酯200 μL,涡旋混合1 min,10 000 r/min离心10 min,分离上清液,残渣再次加入上述溶剂200 μL重复上述操作,萃取3次,合并上清液,用氮气吹干,用50 μL乙腈复溶,离心1 min,取上清液。
2.5.2标准曲线与回收率称取空白大鼠粪便约40 mg于7个离心管中,分别加入混合对照品系列溶液适量,使各组分的浓度为10~400 μg/mL,混匀后,各加入1 200 μL水匀浆,吸取匀浆100 μL于另一离心管中,加入200 μL乙酸乙酯,涡旋混合5 min,10 000 r/min离心10 min,分离上清液,残渣再次加入乙酸乙酯200 μL重复萃取3次,合并上清液,用氮气吹干,50 μL乙腈复溶,取上清液,按“2.3”项下色谱条件检测。结果,各组分在10~400 μg/mL浓度范围内与峰面积呈线性关系,左旋紫草素标准曲线方程Y=91.768X+759.666(R2=0.997 6),乙酰紫草素标准曲线方程Y=383.99X-1 488.3(R2=0.993 7),β,β′-二甲基丙烯酰紫草素:Y=725.13X-1 536.4(R2=0.993 2),最低检测浓度为6 μg/mL。经测定左旋紫草素、乙酰紫草素、β,β′-二甲基丙烯酰紫草素的平均提取回收率分别为(78.2±2.5)%、(82.0±2.9)%和(79.5±3.5)%。
2.6大鼠血浆及粪便中3种萘醌类成分的含量测定
2.6.1收集血浆及粪便取正常SD大鼠20只,随机分为给药组和空白对照组,每组10只,同一条件饲养,两组动物均禁食不禁水12 h。给药组按0.024 g/kg体质量灌胃给予新疆紫草药提取物0.5% CMC-Na溶液混悬,对照组给予0.5% CMC-Na溶液混悬,分别于给药前及给药后15、30 min及1、2、3、4、6、8、12、24、36、48 h 从眼底静脉取血,分离血浆。同时收集给药前及给药后0~12、12~36、36~48、48~72 h的粪便,-20℃冰箱冷冻保存。
2.6.2血浆样品中药物含量的测定血浆样品按“2.4”项下方法处理,按照“2.3”项下色谱条件检测。结果大鼠灌胃后0~48 h内,血浆样品中均未检测到紫草萘醌类成分的特有峰。
2.6.3粪便样品中药物含量的测定称取收集的粪便样品40 mg,各加入1 200 μL水匀浆,吸取匀浆100 μL于另一离心管中,加入200 μL乙酸乙酯,涡旋混合5 min,10 000 r/min离心10 min,分离上清液,残渣再次加入乙酸乙酯200 μL重复萃取3次,合并上清液,用氮气吹干,50 μL乙腈复溶,取上清液,按“2.3”项下色谱条件检测,由标准曲线计算各组分的浓度。结果0~12、12~24、24~28 h粪便中左旋紫草素绝对回收量分别为23.61、38.28、4.13 mg;乙酰紫草素分别为7.20、70.78、11.53 mg;β-β′二甲基丙烯酰紫草素分别为46.32、60.15、16.88 mg。
3讨论
关于紫草萘醌类成分的体内药物浓度测定,主要采用单一成分的测定方法。孙东晓等[7]采用衍生化高效液相色谱法研究了静脉注射乙酰紫草素在大鼠体内的药动学;李慧义等[8]给予大鼠腹腔注射左旋紫草素,收集到的胆汁和血样分别经β-葡萄糖醛酸酶水解后,采用RP-HPLC/DAD方法初步考察了紫草素的体内分布,初步检测到大鼠的胆汁和尿样中分别有10种代谢物。Zhou等[9]采用氧化燃烧炉技术处理小鼠灌胃给予3H-β,β-二甲基丙烯酰紫草素后的组织、粪样(尿样直接加闪烁液测定),用HPLC法分离后用液闪计数仪测定其放射性水平,研究了小鼠体内的组织分布与排泄情况。结果显示,3H-β,β-二甲基丙烯酰紫草素在胃肠道放射性浓度最高,肝脏、肺、肾脏、心脏等组织内放射性浓度次之,骨骼肌、脊髓、脑中分布较少。小鼠灌服 3H-β,β-二甲基丙烯酰紫草素后36 h从粪中收集到给药总放射剂量的72.87%,从尿液中回收到6.81%,粪、尿液中回收到(79.68±0.18)%,血浆中未检测到原形药物。说明3H-β,β-二甲基丙烯酰紫草素在小鼠体内分布广,排泄较完全,主要以原形药物经粪便排泄为主,经肾排泄为辅。在本研究建立的色谱条件下,给予大鼠灌胃紫草提取物后0~48 h,大鼠血浆和尿液中未检测到紫草萘醌类成分,而粪便检测到了紫草萘醌类的3个主要成分,该结果与田慧芳等[10]报告的结果基本一致。说明紫草萘醌类成分在体内分布广,与内源性物质结合呈结合状态,以游离状态分布在血液中的量相对较少,结合态的萘醌类很快被代谢后经粪便排泄。
参考文献:
[1]李国英,买尔旦·马合木提.紫草在新疆的分布及研究进展[J].新疆医科大学报,2009,32(4):386-388.
[2]谢冰芬,冯公侃,黄河,等.天然紫草萘醌类化合物及其衍生物的抗瘤作用研究[J].中国药理学通报,2006,22(4):505-507.
[3]徐新刚,王宝珍,孙志蓉,等.新疆紫草的主要化学成分[J].吉林大学学报(理学版 )2010,48(2):319-322.
[4]朱梦媛,王汝冰,周文,等.紫草素及其衍生物抗肿瘤作用研究进展[J].药学学报,2012,47(5):588-593.
[5]买尔旦·马合木提,刘燕,尼加提·热合木.新疆紫草提取物对D-氨基半乳糖致小鼠急性肝损伤的保护作用[J].中国中药杂志,2006,31(19):1646-1649.
[6]米克热帕·阿布都买买提,买尔旦·马合木提,古丽仙·胡加,等.新疆紫草止血作用研究[J].时珍国医国药,2010,21(11):2889-2590.
[7] 孙东晓,田慧芳,孟志云,等.[3H]-乙酰紫草素在小鼠体内的组织分布、排泄及血中分布研究 [J].军事医学科学院院刊,2008,32 (8):348-352.
[8]李慧义,罗淑荣,周同惠,等.左旋紫草素体内代谢研究[J].中国药学,1999,8(3):148-151.
[9]Zhou JY,Dou GF. Tissue distribution and excretion of radioactivity in mice after intragastric administration of 3H-beta, beta-dimethylacrylshikonin[J]. J Isotopes, 2006,32(3): 45-49.
[10]田慧芳,孙东晓,孟志云,等.小鼠灌胃3H-β,β-二甲基丙烯酰紫草素的组织分布与排泄研究[J].同位素,2008, 21(4):209-214.
(本文编辑施洋)
基金项目:天然药物及仿生药物国家重点实验室(北京大学)开放研究课题(20130215)
作者简介:古力扎努·阿合买提(1989-) ,女(维吾尔族),在读硕士,研究方向:中药民族药基础研究。 通信作者:买尔旦·马合木提,男(维吾尔族),主任药师,教授,硕士生导师,研究方向:中药民族药开发;E-mail:maierdan60@sina.com。
中图分类号:R914
文献标识码:A
文章编号:1009-5551(2016)08-1012-05
doi:10.3969/j.issn.1009-5551.2016.08.019
[收稿日期:2016-04-05]
Determination of three naphthaquinone concentration from Arnebia euchroma (Royle) Johnst in rats plasma and faeces by HPLC methods
Gulizhanu Ahemaiti1, Maierdan Mahemuti2
(1CollegeofPharmacy,XinjiangMedicalUniversity;2AffiliatedTumorHospitalofXinjiangMedicalUniversity,Urumqi830011,China)
Abstract:ObjectiveTo establish a HPLC method for determine of three contents of naphthaquinone from Arnebia euchroma (Royle) Johnst in rats plasma and faeces. MethodsCOSMOIL C18-AR-Ⅱ (4.6 mm×250 mm, 5 μm), column was used with acetonitrile (A) -0.15% formic acid (B)(v/v)as mobile phase by gradient eluting with the flow rate of 1.0 mL/min for 35 min,and the column temperature was maintained at 25℃, detecting wavelength at 274 nm. ResultsThe linear range in plasma of shikonin, acetylshikonin and β-β, -dimethylacrylshikonin were between 0.40 to 6.40 μg, 25.0 to 600.0 μg and 25.0 to 600.0 μg, respectively. The recoveries of these ingredients correspondingly were between 97.78 to 100.1%, 100.1 to100.2% and 97.78 to100.1%. The RSD of intra-day and inter-day were less than 1.5%. The linear range in faeces of shikonin, acetylshikonin and β-β, -dimethylacrylshikonin were between 10-400 μg/mL with the average recoveries of (78.2±2.5)%, (82.0±2.9)% and (79.5±3.5)%, respectively. Absolute recovery amount of Naphthoquinone in feces as: shikonin 0-12 h:23.61 mg,12-24 h:38.28 mg,24-48 h:4.13 mg; acetylshikonin 0-12 h:7.20 mg,12-24 h:70.78 mg, 24-48 h:11.53 mg and β-β, -dimethylacrylshikonin 0-12 h:46.32 mg, 12-24 h:60.15 mg, 24-48h:16.88 mg. ConclusionThe established method has a good selectivity and reproducibility for the determination of naphthaquinone from Arnebia euchroma (Royle) Johnst in plasma and faeces of rats.
Keywords:Arnebia euchroma (Royle) Johnst; shikonin; acetylshikonin; β-β, -dimethylacrylshikonin; HPLC; rat plasma; rat faece
