酶法产L-丙氨酰-L-谷氨酰胺重组大肠杆菌pepD/pepN基因的敲除及其发酵优化
2016-07-21刘沛沛张震宇孙付保周豪
刘沛沛,张震宇,孙付保,周豪
(江南大学 工业生物技术教育部重点实验室,糖化学与生物技术教育部重点实验室,江苏 无锡,214122)
酶法产L-丙氨酰-L-谷氨酰胺重组大肠杆菌pepD/pepN基因的敲除及其发酵优化
刘沛沛,张震宇,孙付保,周豪
(江南大学 工业生物技术教育部重点实验室,糖化学与生物技术教育部重点实验室,江苏 无锡,214122)
摘要L-丙氨酰-L-谷氨酰胺(L-alanyl-L-glutamine, L-Ala-L-Gln),是目前国内外公认的L-谷氨酰胺载体,在临床医学和营养学等领域有广泛应用。为了减少丙谷二肽在生物酶法生产过程中菌体对其降解,利用λ Red同源重组系统,敲除大肠杆菌中肽酶D和氨肽酶对应的编码基因。与野生型菌株相比,双敲除突变菌株的全细胞酶活提高了0.29倍。对该菌株进行摇瓶发酵优化,得到最优培养基为(g/L):葡萄糖 12、酵母提取物 10、胰蛋白胨 10、(NH4)2SO4 1、KH2PO4 3、K2HPO4 1、MgSO4 0.2;最优发酵温度为27 ℃ ;最佳反应条件为:Gln 200 mmol/L 、Ala-Ome·HCl 200 mmol/L,反应pH 8.5,反应温度25 ℃。在最优条件下发酵36 h,丙谷二肽产量达到了14.51 g/L 反应液,是优化前的4.74倍。
关键词L-丙氨酰-L-谷氨酰胺;大肠杆菌;生物酶法;基因敲除;λ Red同源重组;发酵优化
L-谷氨酰胺(L-Glutmine,L-Gln)是一种条件必需氨基酸[1],通常可由肌肉等组织大量合成,当人体处于外伤、手术或感染等应激或病理状态时,Gln的代谢加快,内源合成的Gln不能满足机体的需要,此时必须外源补充[2-3]。Gln具有维持胃肠黏膜正常组织结构[4]、提高机体抗氧化能力、增强机体免疫功能、抗肿瘤及抗抑郁性等重要生理功能[2];在畜禽生产方面,适量的Gln可显著改善断奶仔猪腹泻[5-6],减缓肉仔鸡老化速度并减少疫苗免疫对其的伤害[7]。由于Gln单独存在时,水溶性差(35 g/L, 20 ℃ ),性质不稳定,高温易产生有毒的焦谷氨酸和氨[8],限制了其在临床上的广泛使用。L-丙氨酰-L-谷氨酰胺(L-alanyl-L-glutamine,L-Ala-L-Gln)简称丙谷二肽,是国内外公认的L-Gln载体,因其具有水溶性好(586 g/L, 20 ℃ )、稳定性强、在体内能被很快酶解吸收等优点,使得采用丙谷二肽进行静脉注射可以安全、有效、定量地提供Gln[3]。
目前丙谷二肽的生产方法主要为化学合成法[9],普遍存在成本高、合成过程复杂、合成条件苛刻、所用试剂有毒、易生成副产物等缺陷,不适合大规模工业生产。然而,生物酶法生产丙谷二肽因其具有成本低、污染少、产物得率高等优势,成为近年来的研究热点。2007年,KAZUHIKO T等人[10]在Bacillussubtilis168中发现了L-氨基酸连接酶(Lal),此酶能够催化游离的L-丙氨酸和L-谷氨酰胺合成丙谷二肽,利用大肠杆菌过表达Lal,发酵47 h后,丙谷二肽产量达到24.7 g/L。2011年,YOSHINORI H等[11]克隆表达了SphingobacteriumsiyangensisAJ2458中的α-氨基酸酯酰转移酶(SAET),该酶能以L-丙氨酸甲酯盐酸盐(L-Ala-OMe·HCl)和L-Gln为底物催化合成丙谷二肽,且酶的专一性较强,副产物较少。2013年,YOSHINORI H等[12]利用重组大肠杆菌过表达SAET,实现了大规模生物酶法催化生产丙谷二肽,在25 L发酵规模下,丙谷二肽浓度达到320 mmol/L。
本实验室率先在国内开展生物酶法生产丙谷二肽的研究,将α-氨基酸酯酰转移酶基因(saet基因)置于组成型启动子的控制下,构建出了不需添加诱导剂,能催化生产丙谷二肽的重组大肠杆菌[13]。 本文在本实验室已构建的SAET生产菌株的基础上,敲除大肠杆菌中肽酶D和氨肽酶的编码基因(pepD、pepN),在摇瓶水平上探讨pepD、pepN基因双敲除对SAET表达的影响,并对重组大肠杆菌催化生产丙谷二肽进行了发酵优化。
1材料与方法
1.1材料
1.1.1菌株和质粒
本试验所用菌种及质粒如表1所示。
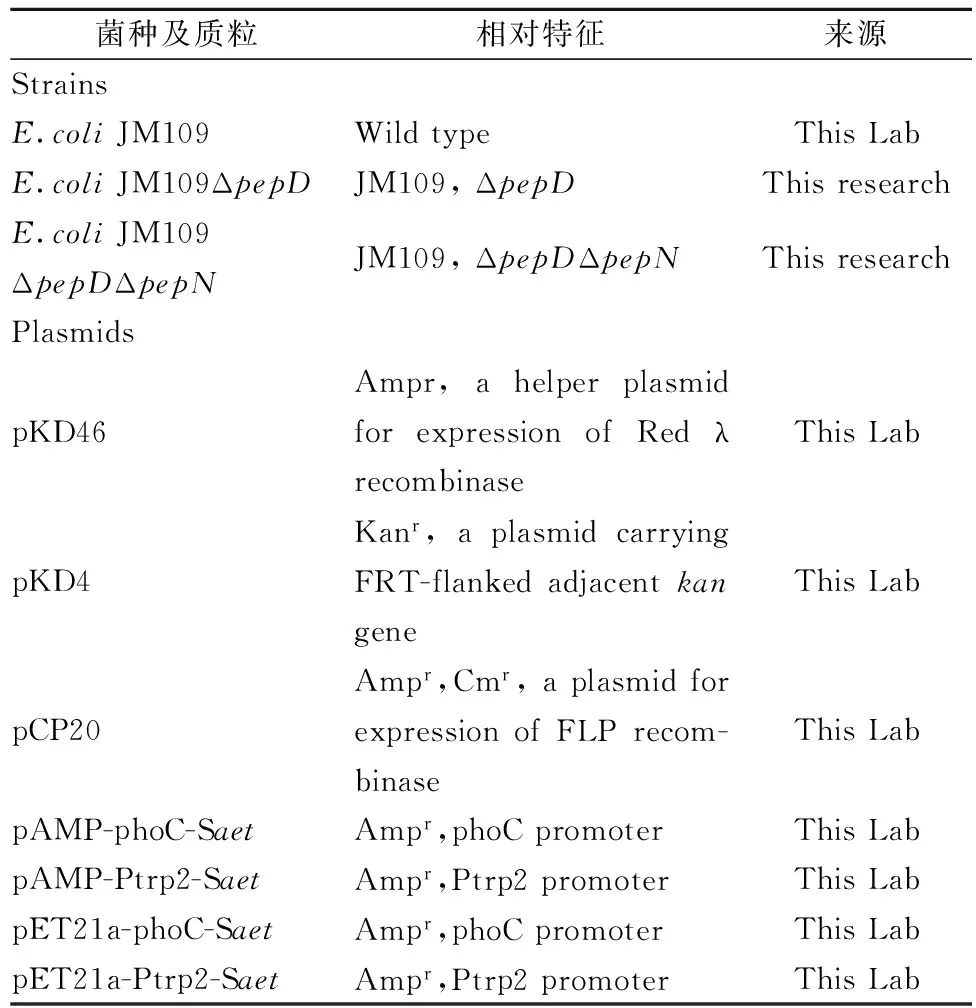
表1 所用菌种、质粒及其来源
1.1.2酶和试剂
TaqDNA聚合酶、PfuDNA聚合酶、1 kb DNA Ladder、质粒小量提取试剂盒、DNA胶回收试剂盒、胰蛋白胨均购自生工生物工程(上海)有限公司;DpnI快切酶、DL5000 DNA Marker及DL10000 DNA Marker购自大连宝生物公司(Takara)公司;L-丙氨酰-L-谷氨酰胺购自TCI公司;L-谷氨酰胺购自国药集团;L-丙氨酸甲酯盐酸盐购自萨恩化学技术有限公司;邻苯二甲醛购自Sigma公司。其他试剂均购自国药集团化学试剂有限公司。
1.1.3 引物
所用引物均由生工生物工程(上海)有限公司合成。敲除及鉴定用引物序列见表2。
1.1.4 培养基
LB培养基(g/L):胰蛋白胨 10,酵母提取物 5,NaCl 10,pH 7.0;固体培养基添加1.8% 琼脂粉。
初始发酵培养基(g/L):葡萄糖20,酵母抽提物10,胰蛋白胨10,(NH4)2SO45,KH2PO43,K2HPO41,MgSO40.5。

表2 敲除和鉴定用引物
The underlined sequence represents complementary to the sequence on the both ends of theKangene
1.2方法
1.2.1基因敲除用打靶片段的制备
以pKD4为模板,分别以DP1和DP2、NP1和NP2为引物,Taq和PfuDNA聚合酶以1∶1的比例混匀进行PCR扩增,DpnI处理后对PCR产物进行切胶回收,以胶回收产物分别作为打靶片段敲除大肠杆菌JM109的pepD基因和JM109(ΔpepD)的pepN基因。
1.2.2大肠杆菌pepD、pepN基因的敲除
大肠杆菌JM109中pepD和JM109(ΔpepD) 中pepN基因的敲除方法参见文献[14],验证用菌落PCR的引物对分别为DY1和DY2、NY1和NY2。
1.2.3细胞培养条件
接种1环重组大肠杆菌至装有30 mL LB液体培养基的250 mL摇瓶中,30 ℃、220 r/min振荡培养10 h,按4%接种至装有25 mL发酵培养基的250 mL摇瓶中,25 ℃、220 r/min振荡培养36 h。
1.2.4完整细胞生产丙谷二肽的反应条件
取2 mL菌液,8 000 r/min离心5 min,去上清液,用等体积的生理盐水洗涤细胞,用上述离心方法再次离心,弃上清,留下的菌体作为催化底物反应合成丙谷二肽的粗酶源,加入500 μL含有50 mmol/L Gln及50 mmol/L Ala-Ome·HCl、pH 8.0的底物溶液,25 ℃ 反应1.5 h,12 000 r/min离心5 min终止反应,取50 μL反应上清液用于丙谷二肽浓度检测。
1.2.5 α-氨基酸酯酰转移酶全细胞酶活的测定
取2 mL菌液,8 000 r/min 离心5 min,去上清,用等体积的生理盐水洗涤细胞,用上述方法再次离心,弃上清,留下的菌体直接作为催化底物反应的酶源,加入500 μL含有200 mmol/L Gln及200 mmol/L Ala-Ome·HCl 、pH 8.0 的硼酸-NaOH缓冲液(0.1 mol/L),25 ℃ 反应5 min,12 000 r/min离心5 min 终止反应,取50 μL反应上清测定丙谷二肽浓度。1个单位酶活定义为:1 min 内生成1 μmol 丙谷二肽所需酶量,单位为U;全细胞酶活定义为单位体积发酵液每克干菌体的酶活,单位为U/DCW,其中细胞干重DCW(g/L)=0.54×OD600。
1.2.6丙谷二肽的检测
柱前衍生:取50 μL标准样品或反应液,加入200 μL 甲醇、200 μL 0.1 mmol/L 四硼酸钠、50 μL衍生剂(20 mg邻苯二甲醛,1.8 mL甲醇,200 μL 0.1 mmol/L四硼酸钠,20 μL β-巯基乙醇,混匀)混匀,于37 ℃ 水浴25 min,加入500 μL 流动相,过0.22 μm有机系膜。
HPLC检测色谱条件:色谱柱 SB-AQ C18(4.6 mm×250 mm, 5 μm);柱温 35 ℃ ;流动相:磷酸缓冲液(12.5 mmol/L , pH 7.2) ∶乙腈 = 9∶1;流速 1 mL/min;激发波长 338 nm,发射波长 450 nm。
1.2.7发酵优化
(1)发酵培养基组分的单因素优化:包括葡萄糖的浓度、硫酸铵的浓度、有机氮源(酵母提取物∶胰蛋白胨 = 1∶1)的浓度以及硫酸镁的浓度。
(2)发酵培养基组分的正交实验:根据单因素优化结果,选取葡萄糖、硫酸铵、有机氮源(酵母提取物∶胰蛋白胨 = 1∶1)和硫酸镁4个因素,按L9(34)正交表实验。
(3)发酵温度优化:发酵温度分别为23、25、27、29、31、33 ℃ 。
(4)反应条件优化:优化了反应pH (7.5、8.0、8.5、9.0)、反应温度(20、25、30、35 ℃ )及两种底物浓度配比(丙氨酸甲酯盐酸盐浓度为200 mmol/L时,谷氨酰胺的添加浓度为50、100、150、200、250 mmol/L)。
2结果与分析
2.1大肠杆菌中pepD和pepN基因的敲除
据报道,在大肠杆菌中,分别由pepA、pepB、pepD、pepN编码的4种肽酶对二肽具有广谱降解活性[15]。一方面,KAZUHIKO T等人通过构建大肠杆菌pepA、pepB、pepD、pepN的单缺失型、双缺失型及多缺失型突变菌株并加入丙谷二肽培养,探究单个或多个二肽酶和氨肽酶的失活对丙谷二肽降解的影响,结果表明,这些突变菌株中的丙谷二肽残留量较野生菌都有不同程度的提高[10],说明这些肽酶对丙谷二肽都有不同程度的降解活性。另一方面,pepD编码的肽酶D是一种二肽酶,其只对二肽有降解活性[16],pepN编码的氨肽酶是大肠杆菌中主要的氨肽酶[17],因此本实验首先考虑在大肠杆菌催化生产丙谷二肽的过程中,敲除pepD、pepN基因,以期减少宿主大肠杆菌对产物丙谷二肽的降解。本文采用λ Red同源重组系统先后敲除大肠杆菌JM109中的pepD、pepN基因,构建双缺失型突变菌株JM109(ΔpepDΔpepN),作为SAET表达的宿主菌株。
首先分别制备pepD和pepN基因敲除的打靶片段。以pKD4为模板,分别以DP1和DP2、NP1和NP2为引物对,扩增出一段两端分别与pepD、pepN基因两端序列同源的,中间为Kan抗性基因的DNA片段,经0.8%的琼脂糖凝胶电泳检测,片段大小与理论值1601 bp、1572 bp一致(图1),经过DpnI处理及胶回收后,所得的产物即可作为JM109中pepD、pepN基因敲除用打靶片段。
A-pepD基因敲除用打靶片段;B-pepN基因敲除用打靶片段图1 基因敲除用打靶片段的电泳分析Fig.1 Electrophoresis analysis of PCR products
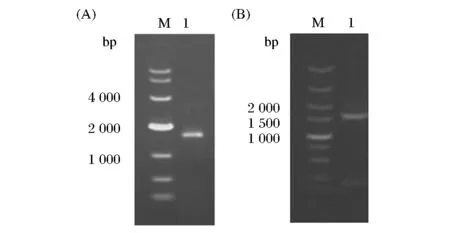
将打靶片段分别电转入含有pKD46的大肠杆菌电转感受态细胞,涂布Kan/LB平板。挑选平板上长出的单菌落做菌落PCR鉴定,分别以DY1和DY2、NY1和NY2为引物,电泳检测PCR产物大小。若以大肠杆菌野生型菌株为模板时,扩增出的条带理论大小分别为1 458 bp、2 859 bp;若Kan抗性基因替代pepD、pepN基因整合到大肠杆菌的基因组上时,扩增出的条带理论大小为1 589 bp、1 835 bp。通过平板筛选及菌落PCR鉴定(图2)可以证明pepD、pepN基因被成功敲除。再将质粒pCP20分别转化至阳性重组子中,通过42 ℃ 高温诱导完成Kan抗性基因的消除及pKD46与pCP20质粒的丢失。并分别以DY1和DY2、NY1和NY2为引物做菌落PCR,以进一步确认Kan抗性基因的消除。若Kan抗性基因从基因组上消除,扩增出的条带理论大小分别为236 bp、441 bp(图3)。从图中可以看出,各个条带的大小均与理论值一致,即成功敲除大肠杆菌JM109中pepD和pepN基因,构建了突变菌株JM109(ΔpepDΔpepN)。
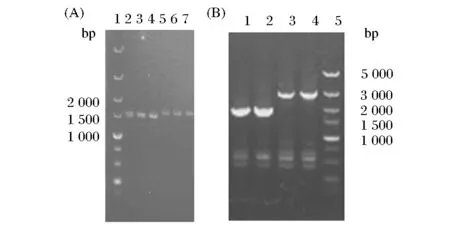
图2 大肠杆菌JM109野生型菌株及基因缺失型菌株PCR电泳图谱Fig.2 Identification of E.coli JM109 gene knockout by colony PCR(A)泳道1:DL5000 DNA Marker;泳道2、3、4:以JM109野生型菌株为模板的菌落PCR;泳道5、6、7:以JM109(ΔpepD)为模板的菌落PCR(B)泳道1、2:以JM109(ΔpepDΔpepN)为模板的菌落PCR;泳道3、4:以JM109野生型菌株为模板的菌落PCR;泳道5:DL5000 DNA Marker
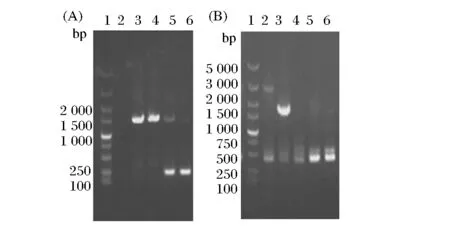
图3 大肠杆菌JM109基因缺失型菌株及Kan抗性消除菌株PCR电泳图谱Fig.3 Identification of the elimination of Kan resistance gene by colony PCR(A)泳道1:DL5000 DNA Marker;泳道2:空白对照;泳道3、4:以JM109(ΔpepD)为模板的菌落PCR;泳道5、6:以去除Kan抗性基因菌株为模板的菌落PCR(B)泳道1:DL5000 DNA Marker;泳道2:JM109野生型菌株为模板的菌落PCR;泳道3:以JM109(ΔpepDΔpepN)为模板的菌落PCR;泳道5、6:以去除Kan抗性基因菌株为模板的菌落PCR
2.2敲除菌与原菌全细胞酶活比较
将本实验室已构建的含SAET的质粒载体pAMP-phoC-SAET(图4)分别转化至野生菌和敲除菌中,比较SAET在野生菌和敲除菌中的表达情况。如表3所示,首先,敲除菌较野生菌,其菌体生长较慢,有报道pepA、pepB、pepD、pepN缺失型突变株的生长比野生型菌株慢[18],一些小肽(如含亮氨酸)会抑制大肠杆菌的生长[15]。综合考虑,肽酶缺失导致胞内一些肽类的累积可能抑制了细胞的生长,但目前还不清楚这些肽酶对胞内二肽的整体降解有多大影响[19]。其次,pepD、pepN双缺失型菌株,其全细胞酶活较野生菌提高了0.29倍,原因可能是双突变菌株的生长较野生菌慢,在相同的发酵时间内,其OD600也较低,进而影响了DCW的值;肽酶D和氨肽酶的失活减少了丙谷二肽的分解,使突变菌株的酶活有所提高。实验结果表明,野生菌的pepD和pepN基因的双敲除,对SAET催化产丙谷二肽有一定的正向促进作用。
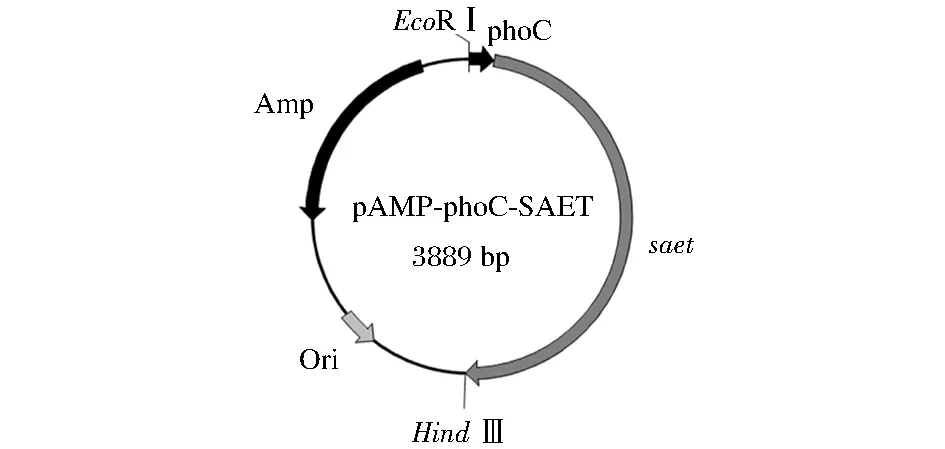
图4 SAET 表达载体Fig.4 SAET expression vector
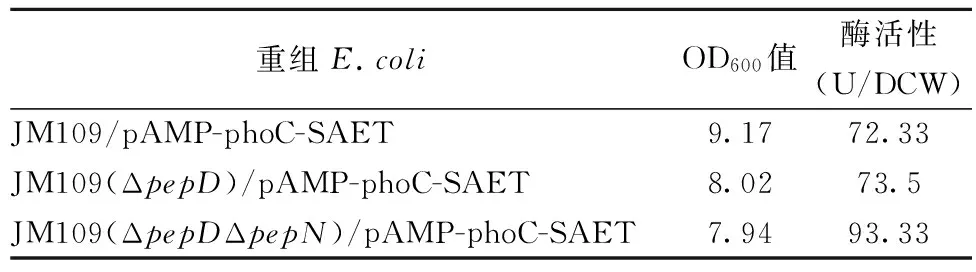
重组E.coliOD600值酶活性(U/DCW)JM109/pAMP-phoC-SAET9.1772.33JM109(ΔpepD)/pAMP-phoC-SAET8.0273.5JM109(ΔpepDΔpepN)/pAMP-phoC-SAET7.9493.33
2.3大肠杆菌生长曲线及种龄的选择
种龄对微生物发酵周期有着重要影响,发酵中一般选择菌种生长的对数中后期为宜。挑取JM109 (ΔpepDΔpepN)/pAMP-phoC-SAET单菌落接种于LB中,菌种OD600与时间的关系如图5所示。菌种接入LB后,4 h开始进入对数期,4~12 h为菌种的对数生长期,12 h后进入稳定期。因此,选择最佳种龄时间为10 h。

图5 重组菌JM109/(ΔpepDΔpepN)/pAMP-phoC-SAET生长曲线Fig.5 The growth curve of recombinant E. coli JM109/(ΔpepDΔpepN)/pAMP-phoC-SAET
2.4重组大肠杆菌发酵曲线及发酵时间的选择
为了确定合适的发酵时间,绘制了重组大肠杆菌的发酵曲线。如图6所示,在发酵36 h之后,丙谷二肽产量没有明显的提高,因此,将重组大肠杆菌的发酵时间定为36 h。

图6 重组菌JM109/(ΔpepDΔpepN)/pAMP-phoC-SAET发酵曲线Fig.6 The fermentation curve of recombinant E. coli JM109(ΔpepDΔpepN)/pAMP-phoC-SAET
2.5发酵培养基单因素优化
2.5.1葡萄糖浓度的优化
在大肠杆菌中,葡萄糖过量会导致溢流效应,葡萄糖经EMP途径后由代谢支路溢出生成乙酸[20],而乙酸的积累会影响菌体的生长和蛋白的表达[21]。对葡萄糖浓度的优化(图7)结果表明,当葡萄糖浓度小于8 g/L时,丙谷二肽产量明显降低;当葡萄糖浓度校大于8 g/L时,丙谷二肽产量逐渐降低,因此,葡萄糖的最佳浓度为8 g/L。
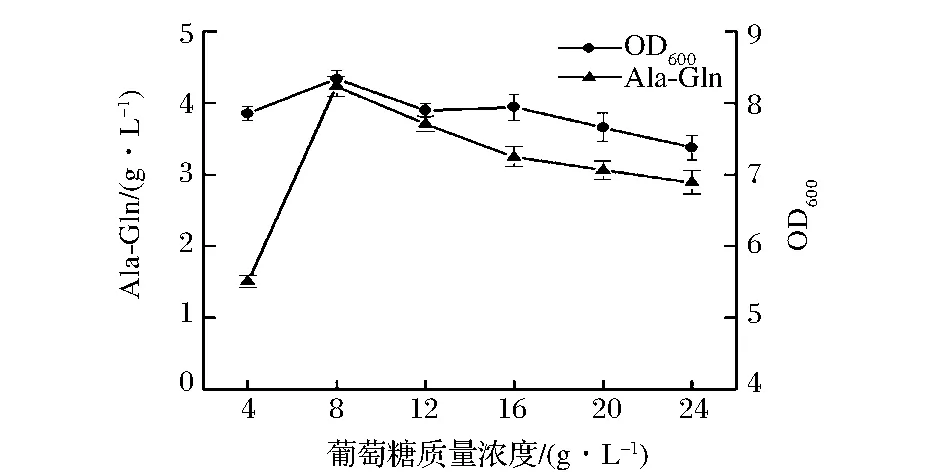
图7 葡萄糖添加量的优化Fig.7 The optimization of carbon source concentration
2.5.2氮源浓度的优化
培养基中的氮源能为微生物提供生长所必需的核苷酸、维生素和矿物质元素等。本文优化了无机氮源硫酸铵的添加浓度,如图8A显示,当硫酸铵添加浓度大于3 g/L时,丙谷二肽产量逐渐降低,因此,硫酸铵添加量定为3 g/L。
与无机氮源相比,有机氮源除含有丰富的蛋白质、肽类、游离的氨基酸以外,还含有少量的糖类,脂肪和生长因子等。据报道,以酵母提取物与胰蛋白胨为有机氮源时,两种氮源以1∶1的比例添加时比单独添加一种有机氮源丙谷二肽产量高[13],因此在优化有机氮源添加量时,以1∶1的比例添加。由图8B可知,当有机氮源为30 g/L时,即酵母提取物与胰蛋白胨添加量分别为15 g/L时,丙谷二肽浓度最高。
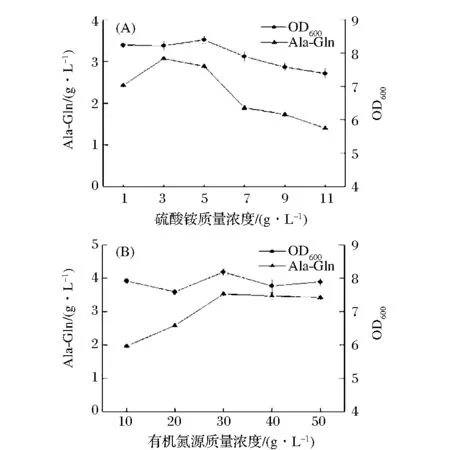
A-无机氮源硫酸铵浓度优化;B-有机氮源浓度优化(酵母提取物∶胰蛋白胨=1∶1)图8 氮源添加量的优化Fig.8 The optimization of nitrogen source concentration
2.5.3硫酸镁质量浓度的优化
Mg2+处于离子状态时,是许多重要酶的激活剂,MgSO4质量不但影响基质的氧化,还影响蛋白质的合成,而硫可以为菌体合成含硫蛋白质提供硫源。本实验考察了MgSO4质量浓度对丙谷二肽产量的影响,结果如图9,当MgSO4质量浓度小于2 g/L时,丙谷二肽产量无明显变化,MgSO4质量浓度为1 g/L时,丙谷二肽产量相对较高;当MgSO4质量补加量超过2 g/L时,丙谷二肽产量明显降低。
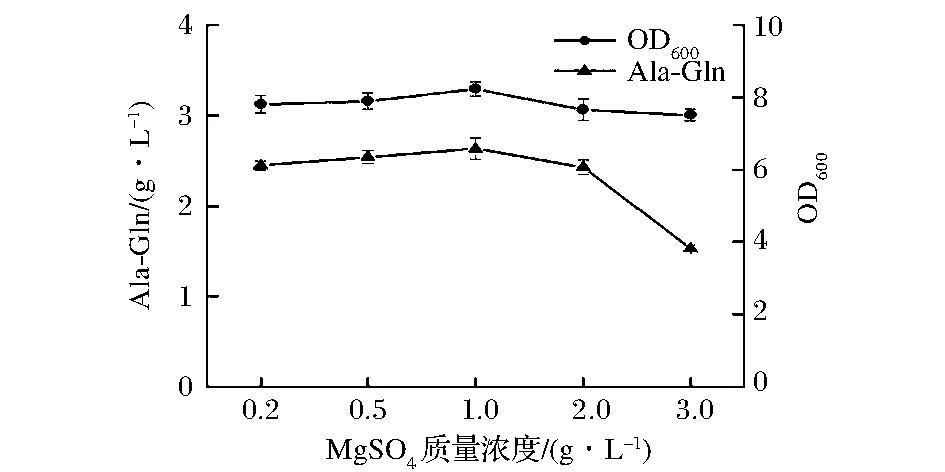
图9 MgSO4质量浓度的优化Fig.9 The optimization of MgSO4 concentration
2.5.4培养基组分的正交实验
根据以上单因素优化结果,考察发酵培养基各组分间的交互作用,选取葡萄糖、硫酸铵、有机氮源(酵母提取物∶胰蛋白胨=1∶1)、MgSO4这4因素做正交实验,每个因素3个水平,本实验选取L9(34)正交表试验,正交实验因素水平表见表4,结果见表5。

表4 正交试验因素水平表
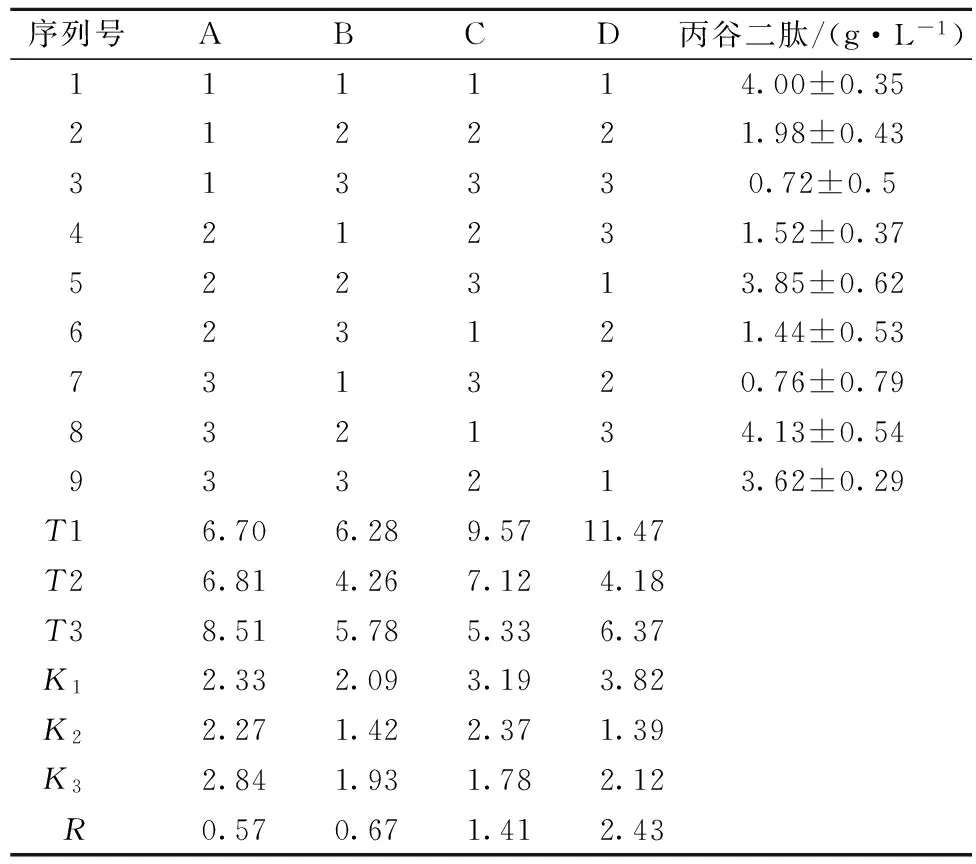
表5 正交实验结果
Ti为各因素同一水平下丙谷二肽产量总和;Ki为Ti平均值;R为极差,表示各因素不同水平下丙谷二肽产量总和的最大差值。
由极差R值可知各因素对丙谷二肽产量的影响顺序为:硫酸镁>有机氮源>硫酸铵>葡萄糖;得到各因素的最佳搭配为:A3B1C1D1。最佳培养基是:12 g/L葡萄糖、1 g/L硫酸铵、10 g/L酵母提取物、10 g/L胰蛋白胨、3 g/L磷酸二氢钾、1 g/L磷酸氢二钾、0.2 g/L硫酸镁。经发酵验证,正交实验所确定的最佳搭配,丙谷二肽产量为5.08 g/L,是优化前(3.06 g/L)的1.66倍。
2.6发酵温度优化
培养温度对菌体生长和目的基因的表达有重要影响,温度除了直接影响发酵过程中各种反应速率外,还通过改变发酵液的物理性质,间接影响菌体的生物合成。如图10所示,发酵温度对丙谷二肽的催化生产有较大影响,当发酵温度为27 ℃ 时,所对应的丙谷二肽浓度最高,当发酵温度高于27 ℃ 时,丙谷二肽产量快速下降。
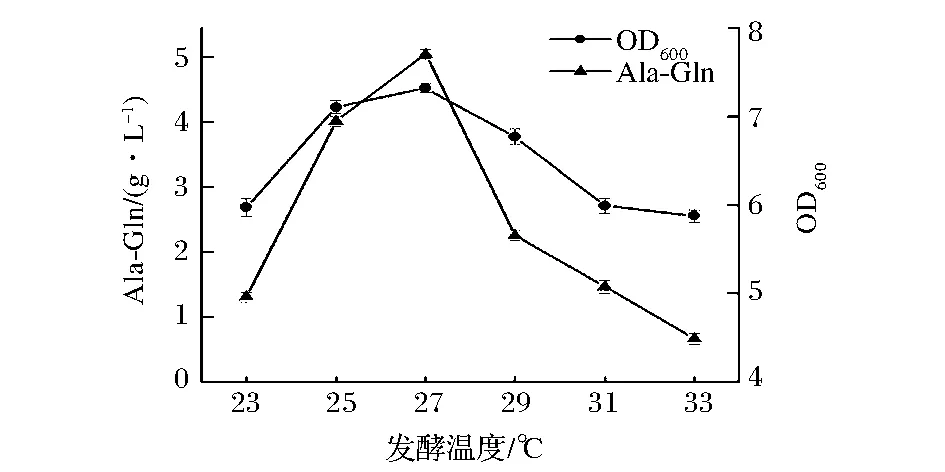
图10 培养温度的优化Fig.10 The optimization of fermentation temperature
2.7酶催化反应条件优化
本实验以发酵菌体为粗酶源,催化L-丙氨酸甲酯盐酸盐和L-谷氨酰胺合成丙谷二肽。酶促反应条件是影响酶活性的重要因素,本实验通过优化催化反应条件,探索产SAET的最适酶促反应条件。
2.7.1反应pH优化
酶的稳定性与其所处环境的pH紧密相关,环境的pH会影响酶分子中相关基团的解离状态,对反应体系pH值的优化结果如图11,反应体系pH值为8.5时,丙谷二肽产量最高,因此将酶促反应的最适pH定为8.5。
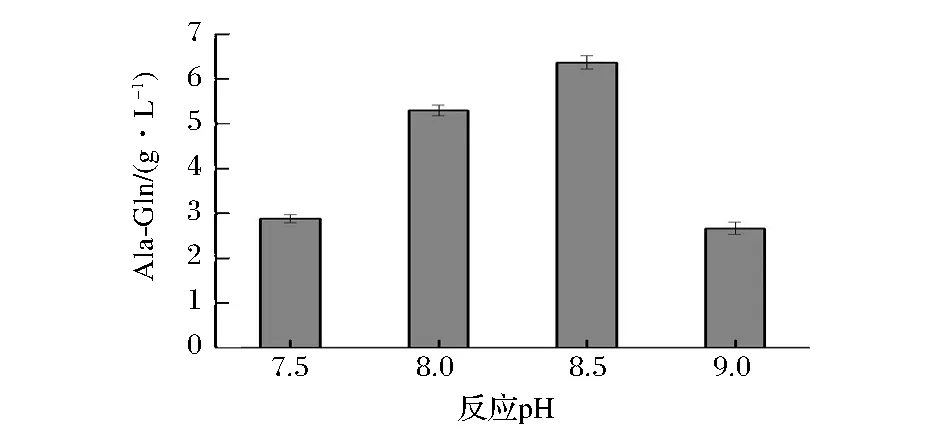
图11 反应液pH的优化Fig.11 The optimization of reaction liquid pH
2.7.2反应温度优化
温度对酶促反应速率的影响主要表现在两个方面,一方面是温度升高时,反应速率加快;另一方面由于酶是蛋白质,随着温度的升高,酶蛋白容易逐渐变性而失活,引起酶反应速率的下降。不同温度对SAET催化生成丙谷二肽的影响结果如图12,当反应温度为25 ℃ 时,丙谷二肽的生成量最高。
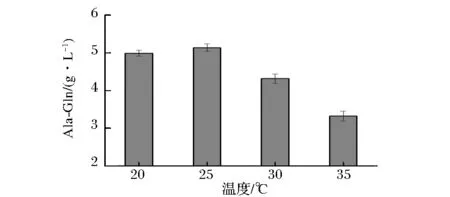
图12 反应温度的优化Fig.12 The optimization of reaction temperature
2.7.3 底物浓度及其配比优化
底物浓度和配比同样对酶促反应有着重要影响,为了确定最适的底物添加量,将Ala-OMe·HCl固定在200 mmol/L,测定不同Gln浓度对反应速率的影响,考察底物浓度对SAET催化产丙谷二肽的影响,结果如图13,随着Gln添加量的加大,丙谷二肽生成量也不断提高,在底物浓度比为200∶200 时,丙谷二肽产量最高。当继续增加Gln的添加量时,丙谷二肽产量不再提高,因此将Ala-OMe·HCl和Gln最适底物浓度及配比定为200∶200。
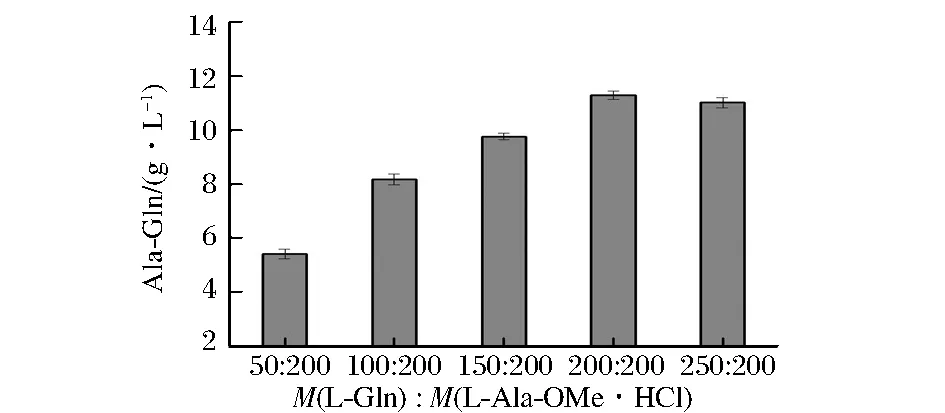
图13 底物配比的优化Fig.13 The optimization of the ratio of the two substrates,AlaOMe·HCl and Gln
3结论
Ala-Gln不仅具有Gln的各种重要的生理功能,且稳定性好,具有重要的临床应用价值。本研究在本实验室成功构建的SAET表达菌株基础上,敲除了宿主大肠杆菌JM109中的肽酶D与氨肽酶对应的编码基因pepD与pepN,构建重组大肠杆菌JM109(ΔpepDΔpepN)/pAMP-phoC-SAET。突变菌株的全细胞酶活较野生菌提高了0.29倍。并进一步优化了重组大肠杆菌发酵生产丙谷二肽的发酵培养基组分、发酵温度及反应条件,在最优条件下,丙谷二肽产量达到了14.51 g/L反应液,是优化前的4.74倍(3.06 g/L),是国内已知生物酶法生产丙谷二肽的最高水平,为丙谷二肽的工业化生产及国产化提供了技术支持。
参考文献
[1]张军民. 条件性必需氨基酸谷氨酰胺研究进展[J]. 中国饲料, 1999(17): 22-24.
[2]贾秀红, 韩琳. 谷氨酰胺的临床应用[J]. 滨州医学院学报, 2008, 31(5): 368-370.
[3]柯远. 丙谷二肽合成工艺改进[D]. 济南:济南大学, 2013.
[4]王岁岁, 梁琨. 谷氨酰胺对早产儿胃肠道屏障及免疫功能的影响[J]. 医学综述, 2010, 16(4): 521-523.
[5]马爽, 王家庆, 王虹玲,等. 谷氨酰胺的生理作用及应用[J]. 安徽农业科学, 2014(26): 9 172-9 173.
[6]邹健, 王康宁. 谷氨酰胺对动物肠道结构和免疫功能的影响[J]. 饲料工业, 2006(3): 17-22.
[7]周荣艳. 饲料中谷氨酰胺及丙氨酰谷氨酰胺的测定[J]. 饲料研究, 2004(1): 30-32.
[8]金辉. 1.谷氨酰胺二肽的合成工艺研究 2.非甾体抗炎镇痛药—扎托洛芬合成研究[D]. 成都:四川大学, 2003.
[9]唐果. N(2)-L-丙氨酰-L-谷氨酰胺二肽的合成与反应研究[D]. 厦门:厦门大学, 2004:15-17.
[10]KAZUHIKO T, SSHIN-ICHI H. Fermentative production ofL-alanyl-L-glutamine by a metabolically engineeredEscherichiacolistrain expressingL-amino acid-ligase[J]. Applied & Environmental Microbiology, 2007, 73(20): 6 378-6 385.
[11]HUBER S, ROOSJE PJ, JANDA J, et al. Gene cloning and characterization of α-amino acid ester acyl transferase inEmpedobacterbrevisATCC14234 andSphingobacteriumsiyangensisAJ2458[J]. Veterinary Immunology & Immunopathology, 2011, 75(11): 2 087-2 092.
[12]HIRAO Y, MIHARA Y, KIRA I, et al. Enzymatic production ofL-alanyl-L-glutamine by recombinantE.coliexpressing α-amino acid ester acyltransferase fromSphingobacteriumsiyangensis[J]. Bioscience, Biotechnology and Biochemistry, 2014, 77(3): 618-623.
[13]何艳春. 产α-氨基酸酯酰基转移酶重组大肠杆菌的构建及发酵优化[D]. 无锡:江南大学, 2015.
[14]DATSENKO K A, WANNER B L. One-step inactivation of chromosomal genes inEscherichiacoliK-12 using PCR products[J]. Proceedings of the National Academy of Sciences of the United States of America, 2000, 97(12): 6 640-6 645.
[15]LAZDUNSKI AM. Peptidases and proteases ofEscherichiacoliandSalmonellatyphimurium[J]. Fems Microbiology Reviews, 1989, 63(3): 265-276.
[16]KLEIN J, HENRICH B, PLAPP R. Cloning and expression of thepepDgene ofEscherichiacoli[J]. Journal of General Microbiology, 1986, 132(8): 2 337-2 343.
[17]DILIP C, DIPANKAR N. PepN is the major aminopeptidase inEscherichiacoli: insights on substrate specificity and role during sodium-salicylate-induced stress[J]. Microbiology, 2004, 149(Pt 12): 3 437-3 447.
[18]HERMSDORF C L, SIMMONDS S, SAUNDERS A. Soluble di‐ and aminopeptidases inEscherichiacoliK-12 dispensible enzymes[J]. European Journal of Allergy & Clinical Immunology, 1979, 13(13): 146-151.
[19]MILLER C G, SCHWARTZ G. Peptidase-deficient mutants ofEscherichiacoli[J]. Journal of Bacteriology, 1978, 135(2): 603-611.
[20]KO YF, BENTLEY WE, WEIGAND WA. An integrated metabolic modeling approach to describe the energy efficiency ofEscherichiacolifermentations under oxygen-limited conditions: Cellular energetics, carbon flux, and acetate production[J]. Biotechnology & Bioengineering, 1993, 42(7): 843-853.
[21]LULI G W, STROHL W R. Comparison of growth, acetate production, and acetate inhibition ofEscherichiacolistrains in batch and fed-batch fermentations[J]. Applied & Environmental Microbiology, 1990, 56(4): 1 004-1 011.
The knockout of genespepDandpepNof recombinantEscherichiacoliproducingL-alanyl-L-glutamine and optimization of its fermentation conditions
LIU Pei-pei, ZHANG Zhen-yu, SUN Fu-bao, ZHOU Hao
(Key Laboratory of Industrial Biotechnology, Ministry of Education,Key Laboratory of Carbohydrate Chemistry and Biotechnology,Ministry of Education, Jiangnan University, Wuxi 214122, Jiangsu, China)
ABSTRACTL-alanyl-L-glutamine is widely recognized as the L-glutamine carrier and applied in the field of clinical medicine and nutrition.In order to interrupt the degradation of proglumetacin dipeptide in E. coli during the biosynthesis process, the λ Red homologous recombination system was employed to knockout the peptidase D and aminopeptidase corresponding coding genes. Compared with wild-type strains, double knockout mutant strain of whole cell enzyme activity increased 0.29 times. The optimized medium composition contained (g/L): glucose 12,yeast extract 10,bacto tryptone 10,(NH4)2SO4 1,KH2PO4 3,K2HPO4 1,MgSO4 0.2. The optimal cultivation temperature is 27 ℃; the most appropriate reaction conditions are: Gln 200 mmol/L, Ala-Ome·HCl 200 mmol/L, reaction pH value is 8.5, reaction temperature is 25 ℃. Finally, after 36 h fermentation under the optimal conditions, the yield is 14.51 g/L reaction liquid which is 4.74 fold that of the initial codition.
Key wordsL-alanyl-L-glutamine; Escherichia coli; enzymatic; gene knockout; λ Red homologous recombination; fermentation optimization
DOI:10.13995/j.cnki.11-1802/ts.201606002
基金项目:国家自然科学基金(30970058);国家自然科学基金(21176106);工业生物技术教育部重点实验室(江南大学)开放课题基金(KLIB-ZR200801)
收稿日期:2016-01-15,改回日期:2016-03-01
第一作者:硕士研究生(张震宇教授为通讯作者,E-mail:zhangzy@jiangnan.edu.cn)。
