Enhanced Electrochemical Performances of LiFePO4/C via V and F Co-doping for Lithium-Ion Batteries
2016-07-05TaotaoZhanChaoLiYunxiaJinShunhuaXiaoCollegeofChemistryandBioengineeringGuangxiKeyLaboratoryofElectrochemicalandMagnetochemicalFunctionalMaterialsGuilinUniversityofTechnologyGuilin541006China
Tao-tao Zhan,Chao Li,Yun-xia Jin,Shun-hua XiaoCollege of Chemistry and Bioengineering,Guangxi Key Laboratory of Electrochemical and Magnetochemical Functional Materials,Guilin University of Technology,Guilin 541006,China
Enhanced Electrochemical Performances of LiFePO4/C via V and F Co-doping for Lithium-Ion Batteries
Tao-tao Zhan,Chao Li,Yun-xia Jin,Shun-hua Xiao∗
College of Chemistry and Bioengineering,Guangxi Key Laboratory of Electrochemical and Magnetochemical Functional Materials,Guilin University of Technology,Guilin 541006,China
(Dated:Received on July 24,2015;Accepted on November 10,2015)
Based on the method of in situ polymerization synthesis combined with two-step sintering process,LiFe1-xVx(PO4)(3-y)/3Fy/C was prepared. The effects of V and F co-doping on the structure,morphology,and electrochemical performances of LiFePO4/C were investigated by X-ray diffraction,Fourier transform infrared spectra,scanning electron microscope,charge/discharge tests,and electrochemical impedance spectroscopy,respectively. The results indicated that the V and F co-doping did not destroy the olivine structure of LiFePO4/C,but it can stabilize the crystal structure,decrease charge transfer resistance,enhance Li ion diffusion velocity,further improve its cycling and high-rate capabilities of LiFePO4/C.
Key words:LiFePO4/C,LiFe1-xVx(PO4)(3-y)/3Fy/C,Electrochemical performances,Diffusion velocity
∗Author to whom correspondence should be addressed. E-mail:xiaoshunhua@glite.edu.cn,Tel.:+86-13457672829,FAX:+86-773-5898551.
I. INTRODUCTION
Nowadays,Li-ion battery has attracted much attentions for its great potential to be used as power sources for electric vehicles and hybrid electric vehicles [1,2]. Among various lithium-ion batteries,the ordered olivine lithium iron phosphate LiFePO4has been recognized as one of the most promising cathode materials for its nontoxicity,low cost,environmental friendliness,long cycling life and high theoretical capacity [3-5]. A lot of researches[6-8]about the synthesis process,structure,electrochemical performance,and applications has been completed,then great progress in physical,chemical properties and synthesis technology had been achieved[9,10]. However,the poor rate capacity of LiFePO4resulting from both the poor electronic conductivity(1×10-10S/m)[11,12]and low Li+diffusion coefficient(1×10-14cm2/s)[13]limits its practical application. So far,various effective methods have been introduced to solve these above problems by using coating particles[14-18],reducing particles sizes[19,20],and doping with guest element[21-23],etc.
Besides carbon coating[24-28]which can effectively improve the electronic conductivity of LiFePO4,cation doping is another effective method to increase the ion electronic conductivity of LiFePO4,such as the doping at Fe-site for LiFe1-xMxPO4(M=V,Ni,Co,Ti,Al,etc.)[29-32]. Similarly,the anion doping can also enhance the electrochemical performances of LiFePO4,such as the rate capability and cycling life[33-41]. Although the study of anion doping was not as full as cation doping,it should not be neglected.
In order to enhance the electrochemical performance of LiFePO4,in this work,the LiFePO4/C sample with V cation and F anion co-doping at different sites was synthesized via the method of in situ polymerization synthesis combined with two-step sintering process. The effects of co-doping with different amount on the structure,morphology,and electrochemical property of LiFePO4/C have been studied in detail.
II. EXPERIMENTS
FePO4/PANI(polyaniline)precursor was prepared by in situ polymerization technology. 100 mL of Fe(NO3)3solution(0.25 mol/L)was added into 200 mL of(NH4)2HPO4(0.125 mol/L)solution with 1 mL aniline,then the pH of the solution was adjusted to 2.1 and stirred for 12 h. The FePO4/PANI precipitate was achieved by filtering and drying under 60◦C vacuum environment for 24 h.
Pristine LiFePO4/C and LiFe1-xVx(PO4)(3-y)/3Fy(x=0.04,y=0.02)powders were synthesized as follows:the stoichiometric amounts of CH3COOLi·2H2O,LiF,NH4VO3,10wt%ascorbic acid and the prepared FePO4/PANI were mixed and milled. After annealing at 350◦C for 5 h under argon atmosphere,10wt%glucose was added and milled again,and then the mixture was annealed at 650◦C for 10 h under argon atmosphere.

TABLE I Lattice parameters of the samples.
The phase identification of powders was conducted with X-ray diffraction measurement(XRD,Panalytical X'Pert PRO MRD,Holland)with a Cu Kα radiation operated at 40 kV and 40 mA. The diffraction data for Rietveld refinement were obtained at 2θ=10◦-70◦,with a step size of 0.013◦. The morphology was evaluated by field emission scanning electron microscopy(FE-SEM S-4800,Hitachi High-Technologies). The FTIR spectra of LiFePO4/C were recorded with a FTIR spectrometer (Nicolet iS10,Thermo Fisher Scientific).
The electrochemical experiments were performed using button cells. The cathode consisted of a mixed material of active materials,carbon black and poly(vinyl difluoride)(PVDF)with mass ratio of 8:1:1 in N-methyl-2-pyrrolidone solvent. Metallic lithium foil was used as the anode electrode. Celgard 2300 polyethylene film was used as separator. 1 mol/L LiPF6solution in ethylene carbonate and dimethyl carbonate(volume ratio of 1:1)was used as electrolyte. The electrochemical performance measurements were carried out with CR2025 coin cells in an argon-filled glove box. Charge/discharge tests were performed using the LAND batteries test system(CT2001A,Wuhan Land Electronic Co.,Ltd.,China)with different current densities. Cyclic voltammetry(CV)were performed on an electrochemical working station(CHI660D,Shanghai Chenhua Co.,Ltd.,China)between 2.5 and 4.2 V.
III. RESULTS AND DISCUSSION
Figure 1 shows XRD patterns of the pristine and V and F co-doped LiFePO4/C. All dominant diffraction lines of samples can be indexed as an olivine phase space group Pnma(JCPDS card No.83-2092),which were consistent with the previous reports[42,43]. Typical diffraction peaks of carbon,vanadium and fluorine can not be found,indicating that the contribution of the carbon was less or the carbon existed in amorphous form,and the elements V and F probably entered into the lattice of LiFePO4rather than form impurities. Both samples had narrow diffraction peaks,which indicated good crystallinity degree. Figure 1(b)shows the magnification of the field of 35◦-36◦,there was a very slight shift to lower angle for the doped sample,it was in line with convention that the diffraction peak with larger unit cell volume would shift towards lower diffraction angles in XRD pattern.
What's more,the crystal lattice parameters were calculated by MDI Jade 5.0,and the results are listed in Table I. As shown,that V and F doping led to slight increase of lattice parameters. The doped sample expanded the b and c by 0.14%and 0.06%respectively,and shrinked a by 0.017%,which indicated that the V and F entered into the crystal structure and caused lattice distortion in the LiFePO4/C unit cell. The volume of the unit cell of the doped sample was slightly larger than that of the pristine LiFePO4/C,which was beneficial to intercalation and deintercalation for Li ion.
The details of the chemical bonding in the LiFe1-xVx(PO4)(3-y)/3Fy/C samples were investigated in the FTIR spectra,as shown in Fig.2. The FTIR spectra exhibited many prominent and narrow multiple vibration bands in the range of 500-2000 cm-1. For two samples,the absorption bands centered at 635.59,966.09,1051.99,1095.55,and 1138.06 cm-1corresponded to the symmetric and asymmetric modes of(PO4)3-groups. In the lower wave number region,the absorptions at 471.57,501.10,548.76,576.39 cm-1were attributed to the symmetric and asymmetric bending modes of P-O bonds[44]. In addition,the absorption at 1630 cm-1can be as-signed to the stretching vibration of the aromatic ring C=C bonds in grapheme. Most importantly,that the peaks of LiFe0.96V0.04(PO4)2.98/3F0.02/C samples shifted slightly to higher band numbers,implying the doping ions entrance and the resulted absorption shift.
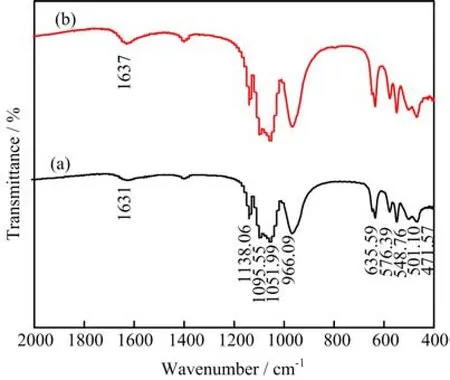
FIG. 2 FTIR spectrum of LiFe1−xVx(PO4)(3−y)/3Fy/C. (a)LiFePO4/C,(b)LiFe0.96V0.04(PO4)2.98/3F0.02/C.

FIG. 3 SEM images of LiFe1−xVx(PO4)(3−y)/3Fy/C samples.(a)x=0,y=0,(b)x=0.04,y=0.02.
SEM images of LiFe1-xVx(PO4)(3-y)/3Fy/C samples are shown in Fig.3. It indicated that the morphology of LiFePO4/C particles was sphere-like similar to that of LiFe0.96V0.04(PO4)2.98/3F0.02/C. It showed that the in situ polymerization synthesis method can effectively control the size and morphology of samples,and the V and F doping did not result in evident influence on the particles morphology. In comparison,LiFe0.96V0.04(PO4)2.98/3F0.02/C sample presented better dispersion and uniformity than that of LiFePO4/C sample.
Rate capabilities of LiFe0.96V0.04(PO4)2.98/3F0.02/C,LiFePO4/C,and LiFe0.96V0.04PO4/C samples from 0.2 C to 10 C are shown in Fig.4. It is shown that the LiFe0.96V0.04PO4/C sample had a higher discharge capacity in comparison with the LiFePO4/C sample at a modest scope. The LiFe0.96V0.04(PO4)2.98/3F0.02/C sample revealed the best rate capability among three samples,especially under high rate conditions. It delivered a discharge capacity of 162.6(0.2 C),143.6 (0.5 C),138.2(1 C),124.3(2 C),94(5 C),76.8(10 C)mAh/g respectively. The capacity of the un-doped sample decreased dramatically with the rate increasing,remained only 40 mAh/g at 10 C,while the co-doped sample showed a capacity of 76.8 mAh/g at 10 C. The superior rate performance of LiFe0.96V0.04(PO4)2.98/3F0.02/C should result from the combined effects of V and F doping on the increasing conductivity of LiFePO4/C.
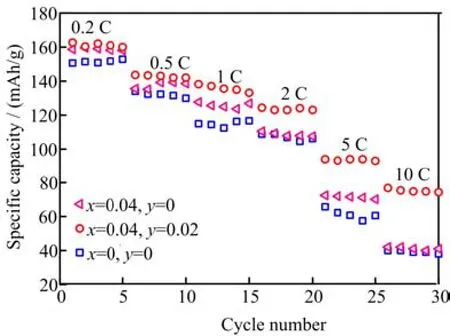
FIG. 4 The rate profiles of LiFe1−xVx(PO4)(3−y)/3Fy/C composites.
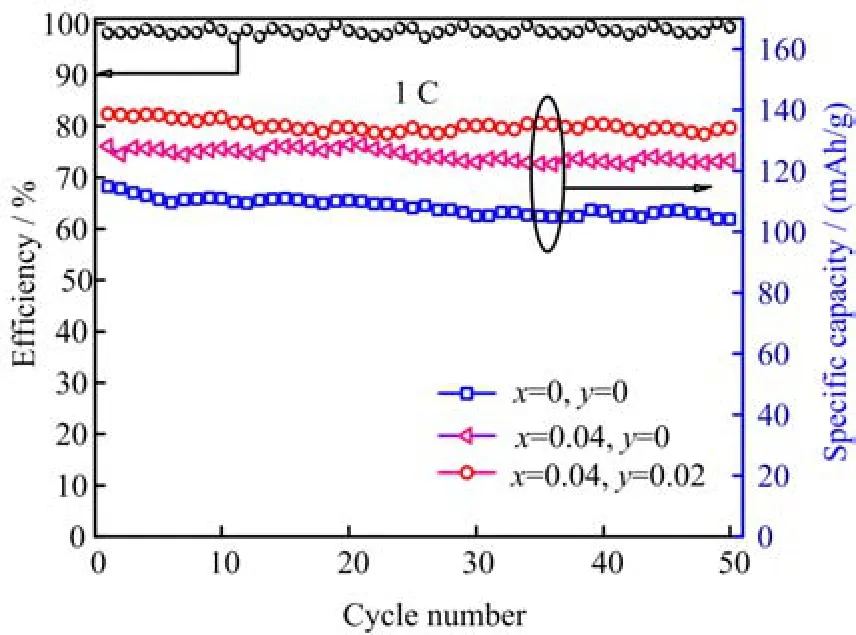
FIG. 5 Cycle performance profiles and coulombic efficiency of LiFe1−xVx(PO4)(3−y)/3Fy/C at 1 C.
Figure 5 shows cycle performances and coulombic efficiency of LiFe1-xVx(PO4)(3-y)/3Fy/C at a current rate of 1 C. It can be observed that LiFe0.96V0.04(PO4)2.98/3F0.02/C displayed a higher capacity retention than that of LiFePO4/C and LiFe0.96V0.04PO4/C after 50 cycles. This indicated that the structural stability of LiFePO4/C was improved by V and F co-doping. The result was consistent with the conclusion about structure investigation. In addition,near 100%coulombic efficiency for LiFe0.96V0.04(PO4)2.98/3F0.02/C samples was achieved,which may be attributed to the fact that multicomponent doping leads to faster ion diffusion rate,and then slower capacity decay.
The Nyquist plots for LiFePO4/C and LiFe0.96V0.04(PO4)2.98/3F0.02/C samples are pre-sented in Fig.6. The high-frequency semicircle reacted to the charge transfer resistance(R1),which was caused by charge transfer across the electrode electrolyte interface,whose diameter was approximately equal to the charge transfer resistance(R1). The straight line at lower frequency presented Li+diffusion resistance in electrode bulk,namely,the Warburg impedance(Zw),which was associated with Li ions diffusion in LiFePO4/C particles. As expected,the R1of LiFePO4/C electrode decreased in the charge/discharge process after V and F co-doping. The results indicate that the impedance of electrochemical reaction is improved by co-doping.
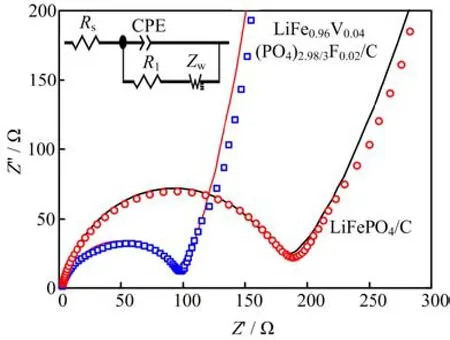
FIG. 6 EIS spectra of LiFe0.96V0.04(PO4)2.98/3F0.02/C and and LiFePO4/C electrodes. Solid lines show the fitting results.
Figure 7 shows the CV curves of the second cycle at different scan rates between 0.1 and 0.6 mV/s within the potential window between 2.5-4.2 V. A couple of redox peaks corresponding to the lithium insertion and formation of LiFePO4/C phase were observed. Obviously,the peak shape of LiFe0.96V0.04(PO4)2.98/3F0.02/C was sharper and the peak separation was narrower than that of LiFePO4/C.
At the same time,the diffusion coefficients of lithium ions(DLi+)in both samples were also calculated by CV,which was widely used to study the oxidation-reduction and phase transformation processes in electrode reactions and to estimate quantitatively DLi+. The corresponding fitting results are shown in Fig.8,it can be found that the redox peak currents of both LiFePO4/C and LiFe0.96V0.04(PO4)2.98/3F0.02/C electrodes showed a linear relationship with the square root of scan rate. For semi-infinite and finite diffusion,the peak current (ip)was proportional to the square root of the scan rate and could be expressed by the classical Randles-Sevcik equation[45]:
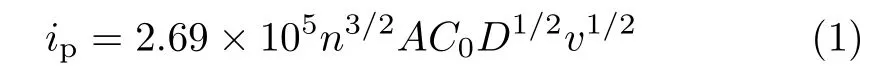
where n is the number of electrons involved in the redox process,A is the electrode area,C0was the molar concentration of lithium ions,D is the diffusion coefficient of lithium ions,and v is the potential scan rate. Based on the equation and the slope of ipvs. v1/2in Fig.8,we achieved the DLi+of co-doping and initial samples during oxidation process were 1.00×10-10and 4.98×10-11cm2/s. Obviously,V and F co-doping increased Li+diffusion coefficient of LiFePO4/C.

FIG. 7 CV curves of LiFePO4/C(a)and LiFe0.96V0.04(PO4)2.98/3F0.02/C(b)at different scan rate.

FIG. 8 Relationship of the peak current(ip)and the square root of scan rate(v1/2).
IV. CONCLUSION
In this work,vanadium and fluorine co-doped LiFePO4/C cathode materials with uniform size and spherical morphology were synthesized via in situ poly-merization method combined with two-step sintering route. The V and F ions were successfully doped into the LiFePO4/C phase. The co-doping of V and F ions can enhance the structural stability,decrease charge transfer resistance,increase electronic conductivity,improve Li ion diffusion velocity,and finally result in eminent cycling and high-rate capabilities of LiFePO4/C. Therefore,it can be concluded that the vanadium and fluorine co-doping was an effective way to improve the electrochemical performances of LiFePO4/C cathode materials for lithium-ion batteries.
V. ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China(No.51364008),the Science and Technology Research Projects of Guangxi institution of higher education(No.2013ZD030),and the Guangxi Natural Science Foundation (No.2014GXNSFAA118046).
[1]M. Armand and J. M. Tarascon,Nature 451,652 (2008).
[2]P. Gibot,M. Casas-Cabanas,L. Laffont,S. Levasseur,P. Carlach,and J. M. Tarascon,Nature Mater. 7,741 (2008).
[3]A. Padhi,K. S. Nanjundaswamy,C. Masquelier,S. Okada,and J. B. Goodenough,J. Electrochem. Soc. 144,A1609(1997).
[4]M. Zhong and Z. Zhou,Solid State Ionics 181,1607 (2010).
[5]T. Nagakane,H. Yamauchi,K. Yuki,M. Ohji,A. Sakamoto,T. Komatsu,H. Honma,M. Zou,G. Park,and T. Sakai,Solid State Ionics 206,78(2012).
[6]J. H Yum,E. Baranoff,S. Wenger,and M. K. Nazeeruddin,Energy Environ. Sci. 4,842(2011).
[7]Y. G. Wang,P. He,and H. Zhou,Energy Environ. Sci. 4,805(2011).
[8]J. Y. Li,W. L. Yao,S. Martin,and D. Vakin,Solid State Ionics 179,2016(2008).
[9]G. Atnold,J. Garche,R. Hemmer,S. Strobele,C. Vogler,and M. Wohlfahrt Mehrens,J. Power Sources 119,247(2003).
[10]V. Srinivasan and J. Newman,J. Electrochem. Soc. 151,A1517(2004).
[11]K. Rissouli,K. Benkhouja,J. R. Ramos-Barrado,and C. Julien,Mater. Sci. Eng. B 98,185(2003).
[12]M. Gaberscek,R. Dominko,and J. Jamnik,Electrochem. Commun. 9,2778(2007).
[13]P. P. Prosini,M. Lisi,and D. Zane,Solid State Ionics 45,148(2002).
[14]R. Dominko,M. Gaberscek,J. Drofenik,M. Bele,S. Pejovnik,and J. Jamnik,J. Power Sources 119,770 (2003).
[15]N. Ravet,Y. Chouinard,J. F. Magnan,S. Besner,M. Gauthier,and M. Armand,J. Power Sources 97,503 (2001).
[16]F. F. Pan,W. L. Wang,D. M. Chen,and W. S. Yan,J. Mater. Chem. 21,14680(2011).
[17]F. F. Pan,W. L. Wang,H. J. Li,and X. D. Xin,Electrochim. Acta 56,6940(2011).
[18]Q. Q. Chang,G. H. Yao,F. F. Pan,and D. M. Chen,Electrochim. Acta 108,211(2013).
[19]W. J. Zhang,J. Power Sources 196,2962(2011).
[20]R. Malik,D. Burch,M. Bazant,and G. Ceder,Nano Lett. 10,A4123(2010).
[21]X. Q. Ou,H. C. Gu,Y. C. Wu,J. W. Lu,and Y. J. Zheng,Electrochim. Acta 96,230(2013).
[22]J. Hong,X. L. Wang,Q. Wang,and F. Omenya,J. Phys. Chem. C 116,20787(2012).
[23]H. Y. Gao,L. F. Jiao,W. X. Peng,and G. Liu,Electrochim. Acta 56,9961(2011).
[24]Z. H. Wang,L. X. Yuan,J. Ma,L. Qie,and L. Zhang,Electrochim. Acta 62,416(2012).
[25]L. Wang,H. Wang,Z. Liu,C. Xiao,and S. Dong,Solid State Ionics 181,1685(2010).
[26]J. Wang,Z. Shao,and H. Ru,Ceram. Int. 40,6979 (2014).
[27]K. Park,S. Schougaard,and J. Goodenough,Adv. Mater. 19,848(2007).
[28]G. Qin,Q. Ma,and C. Wang,Solid State Ionics 257,60(2014).
[29]M. Abbate,S. M. Lala,L. A. Montoro,and J. M. Rosolenb,Electrochem. Solid State Lett. 8,A288 (2005).
[30]Z. H. Wang,Q. Q. Pang,J. K. Deng,and L. X. Yuan,Electrochim. Acta 78,576(2012).
[31]Z. Liu,X. Huang,and D. Wang,Solid State Commun. 147,505(2008).
[32]L. Yang,L. Jiao,Y. Miao,and H. Yuan,J. Solid State Electrochem. 13,1541(2009).
[33]X. Liao,Y. He,and Z. Ma,J. Power Sources 174,720 (2007).
[34]R. Malik,D. Burch,M. Bazant,and G. Ceder,Nano Lett. 10,4123(2010).
[35]F. Lu,Y. C. Zhou,J. Liu,and Y. Pan,Electrochim. Acta 56,8833(2011).
[36]F. Pan and W. L. Wang,J. Solid State Electrochem. 16,1423(2012).
[37]M. Milovi´c,D. Jugovi´c,N. Cvjeti´canin,and D. Uskokovi´c,J. Power Sources 241,70(2013).
[38]D. Wang,H. Li,and Z. Wang,J. Solid State Chem. 177,4582(2004).
[39]B. Wu,Y. Zhang,and N. Li,J. New Mater. Electrochem. Syst. 14,147(2011).
[40]G. Amatucci and N. Pereira,J. Fluor. Chem. 128,243 (2007).
[41]X. Z. Liao,Y. S. He,Z. F. Ma,and X. M. Zhang,J. Power Sources 174,720(2011).
[42]M. Yang and W. Ke,J. Electrochem. Soc. 155,A729 (2008).
[43]T. Teng,M. Yang,and S. Wu,Solid State Commun. 142,389(2007).
[44]A. Salah,P. Jozwiak,and J. Garbarczyk,J. Power Sources 140,370(2005).
[45]J. Dahn,J. Jiang,and L. Moshurchak,J. Electrochem. Soc. 152,A1283(2005).
DOI:10.1063/1674-0068/29/cjcp1507163
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Virtual Screening of Human O-GlcNAc Transferase Inhibitors
- Comparative Theoretical Studies on Several Energetic Substituted Dioxin-imidazole Derivatives
- Controlled Synthesis of PCL/PVP Copolymer by RAFT Method and Its Hydrophilic Block-Dependent Micellar Behaviors
- Epitaxial Growth and Thermoelectric Measurement of Bi2Te3/Sb Superlattice Nanowires
- Morphology and Growth Process of Bat-like ZnO Crystals by Thermal Evaporation
- Investigation of Ultrafast Electronic Transfer Process on Organic/Inorganic Heterojunction by Femtosecond Transient Absorption