Comparative Theoretical Studies on Several Energetic Substituted Dioxin-imidazole Derivatives
2016-07-05MeiZhengXiaohongLiHonglingCuiRuizhouZhangCollegeofPhysicsandEngineeringHenanUniversityofScienceandTechnologyLuoyang471003China
Mei Zheng,Xiao-hong Li,Hong-ling Cui,Rui-zhou ZhangCollege of Physics and Engineering,Henan University of Science and Technology,Luoyang 471003,China
Comparative Theoretical Studies on Several Energetic Substituted Dioxin-imidazole Derivatives
Mei Zheng,Xiao-hong Li∗,Hong-ling Cui,Rui-zhou Zhang
College of Physics and Engineering,Henan University of Science and Technology,Luoyang 471003,China
(Dated:Received on June 06,2015;Accepted on August 06,2015)
The molecular structures,infrared spectra,heats of formation(HOFs),detonation properties,chemical and thermal stabilities of several tetrahydro-[1,4]dioxino[2,3-d:5,6-d′]diimidazole derivatives with different substituents were studied using DFT-B3LYP method. The properties of the compounds with different groups such as -NO2,-NH2,-N3,and -ONO2were further compared. The -NO2and -ONO2groups are effective substituents for increasing the densities of the compounds,while the substitution of -N3group can produce the largest HOF. The compound with -NO2group has the same detonation properties as 1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane,while the compound with -ONO2group has lower detonation properties than those of hexahydro-1,3,5-trinitro-1,3,5-triazine. The nature bond orbital analysis reveals that the relatively weak bonds in the molecules are the bonds between substituent groups and the molecular skeletons as well as C-O bonds in the dioxin rings. The electron withdrawing groups(-NO2,-N3,and -ONO2)have inductive effects on the linkages between the groups and molecular skeletons. In addition,researches show that the electronegativities of the groups are related with the stabilities of the compounds. Considering detonation performance and thermal stability,the 1,5-dinitro-2,6-bis(trinitromethyl)-3a,4a,7a,8a-tetrahydro-[1,4]dioxino-[2,3-d:5,6-d′]diimidazole satisfies the requirements of high energy density materials.
Key words:Density functional theory,Condensed density,Heats of formation,Detonation property,Thermal stability
∗Author to whom correspondence should be addressed. E-mail:lorna639@126.com.
I. INTRODUCTION
The use of imidazole derivatives as high energy density materials(HEDMs)to satisfy both civil and military applications was extensive during the past decades[1-4]. Imidazole rings are relatively stable and rich in C and N atoms,so they are considered as HEDMs when linked with functional groups[5]. Various nitroimidazole derivatives were synthesized and have been investigated experimentally and theoretically. Results show that many of nitroimidazole derivatives meet excellent properties as HEDMs,such as cis-syn-cis-2,6-dioxo-1,3,4,5,7,8-hexanitrodecahydro-1H,5H-diimidazo[4,5-b:4′,5′-e]pyrazine(CL-20). It is polycyclic and is rich in nitro groups,which results in high density(2.07 g/cm3),and good detonation properties[5-7].
The synthesis of a nitrogen-rich imidazole poly-tri-heterocyclic compound,1,5-dinitro-2,6-bis(trinitromethyl)-3a,4a,7a,8a-tetrahydro-[1,4]dioxino-[2,3-d:5,6-d′]diimidazole(DNTNDI)was reported by Wu et al.[8]. DNTNDI is constituted by a 1,4-dioxin ring connecting two methyl-imidazole rings,and all the H atoms on the two methyl-imidazole rings are substituted by nitro groups except the H atoms on the dioxin ring. Experiments show that DNTNDI has an excellent density of 1.929 g/cm3and its oxygen balance is zero,which imply that the moles of gas production and the heat of detonation may reach maximum.
Generally,nitrate ester group(-ONO2)is an organic nitric acid group that can contain enormous explosive force and is often used to modify the mechanical properties[9]. The introduction of an amino group(-NH2)is one of the simplest means to improve the thermal stability of an energetic material[10]. In addition,azido group(-N3)has a high positive heat of formation and can produce more gases in the combustion products,thereby increasing the work capacity of propellant[11]. Considering the importance of the three groups,we wonder what changes the properties of the new energetic materials if the -NO2groups are substituted by -NH2,-ONO2,and -N3groups,respectively.
Heat of formation(HOF)is crucial thermodynamic quantity and is frequently taken to be indicative of the “energy content”of a HEDM[12,13]. Detonation velocity(D)and pressure(P)are the most important ta-rgets of scaling the detonation characteristics of energetic materials. The bond dissociation energy(BDE)of the weakest bond for an explosive molecule is a key factor in many decomposition processes[14],and has been expected to play an important role in the initiation of detonation. Lots of calculations indicate that C-NO2and N-NO2bonds are the weakest bonds in energetic ring molecules and are the first rupture bond during decomposition process. The BDEs are also related to the stabilities and impact/shock sensitivities of HEDMs[15-18]. Therefore,it is necessary to understand the properties and the internal mechanism of DNTNDI by using the theoretical investigations.
In this work,the molecular structures,infrared spectroscopy,HOF,thermal stabilities,and detonation properties are calculated for DNTNDI and the derivatives with -NH2,-ONO2,and -N3groups by using density functional theory B3LYP method. The related properties of these compounds are predicted and their molecular structures are listed in Fig.1.

FIG. 1 Molecular frameworks of DNTNDI and derivatives 1,2,3.
II. COMPUTATIONAL METHOD AND DETAILS
In this work,the theoretical data for DNTNDI and its derivatives compounds 1,2,3(Fig.1)were calculated by using density functional theory(DFT)B3LYP method with 6-311++G∗∗basis set. The DFT method,particularly the B3LYP[19-22]method was testified as a credible approach which not only produces reliable geometries,energies and electron population but also is efficient[23]. All the calculations are performed by Gaussian 03 program package[24].
Infrared Radiation(IR)spectrum is a measurable characteristic of a material which can effectively identify groups and/or ingredients. And it is directly related to the thermodynamic properties of compounds. Thus,the IR spectra for DNTNDI and compounds 1,2,3 were calculated based on optimized molecular structures. Besides,the previous investigations revealed that the calculated vibrational frequencies conform to the experimental frequencies with an appropriate scaling factor[25-28].
The isodesmic reactions were designed to evaluate HOFs of DNTNDI and compounds 1,2,3. Isodesmic reaction method has been widely and successfully used in predicting HOFs[29-32]. The isodesmic reactions remain the number of each kind of formal bond during being performed. For the title compounds,the HOFs were derived from the isodesmic reactions shown in Fig.2.
To calculate the detonation properties of DNTNDI and compounds 1,2,3,solid-phase HOFs(∆Hf,solid)are required. The∆Hf,solidcan be evaluated by the employment of gas-phase HOFs(∆Hf,gas)and heats of sublimation(∆Hsub),as Eq(1):
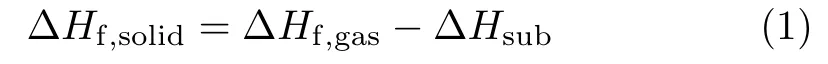
Politzer et al.[33,34]pointed out that the heatsof sublimation of energetic materials containing CHNO atoms are related to their molecular surface areas and electrostatic interaction index. They presented the following relationship:

where A is the 0.001 electrons/bohr3isosurface area of electronic density of the molecule,ν describes the degree of balance between positive and negative potential on the isosurface,andis a measure of variability of the electrostatic potential on the molecular surface. The coefficients a,b and c were determined by Rice et al.,a=2.670×10-4kcal/mol,b=1.650 kcal/mol,and c=2.966 kcal/mol[35]. Furthermore,Politzer et al. [33-35]found that the solid density of energetic materials containing CHNO atoms can be corrected by applying parameterfor the theoretical density:

where M is the molecular weight in g/mol,V is the volume inside a contour of 0.001 electrons/bohr3electron density for the molecule. The volume was computed using a Monte Carlo integration that set 2000 point/bohr3for adequate accuracy. The coefficients β1,β2,and β3are 0.9183,0.0028,and 0.0443,respectively.
The detonation velocity D and pressure P can be estimated by the semi-empirical Kamlet-Jacobs equations [36]:

where N is the moles of gas phase products from per gram of the explosiveis the average molecular mass of these gases,Q is the standard enthalpy change of the detonation from per gram explosive,ρ is the loaded density of explosives.

FIG. 2 Isodemic reactions of compound 1,2,3.
The frontier molecular orbitals(FMO)and the average binding energies(ABEs)for title compounds were calculated using B3LYP/6-311++G∗∗method in order to evaluate the chemical stabilities. ABE is defined as the quotient that the molecular binding energy divides the number of atoms in a molecule. The ABE can be calculated using the equation[37]:

where E(atomi)is the energy of the ith atom,E(molecule)is the total energy of the molecule,n is the number of atoms in the molecule.
In order to understand the stability of each bond in the title compounds,the natural bond orbital(NBO)[38]calculation was performed by using B3LYP/6-311++G∗∗method. Based on the NBO analysis,pyrolysis mechanism and thermal stability of the title compounds were investigated via BDE. The BDE is the difference between the energies of parent molecule and radicals which are from the fission of the molecule [39-41]. The BDE can be calculated by using the following equation[42]:

where R1and R2are the radicals produced by the breaking of R1-R2bond of R1R2compound.
III. RESULTS AND DISCUSSION
A. Molecular geometry
The molecular geometries of the title compounds were optimized by B3LYP/6-311++G∗∗method. For DNTNDI and compounds 2,3,the imidazole rings and the oxygen bridges are generally planar,while the oxygen atoms on dioxin ring are slightly deviated from the plane. Compound 1 looks like a curving boat with imidazole rings and oxygen bridges being not on a plane.
Selected bond lengths of DNTNDI and compounds 1,2,3 are listed in Table I. For comparison,the bond lengths of the normal single bonds(experimental and calculated bond length)are also shown in Table I. Obviously,the calculated bond lengths are very close to the experimental normal single bond lengths. This shows that our calculational method is reliable.
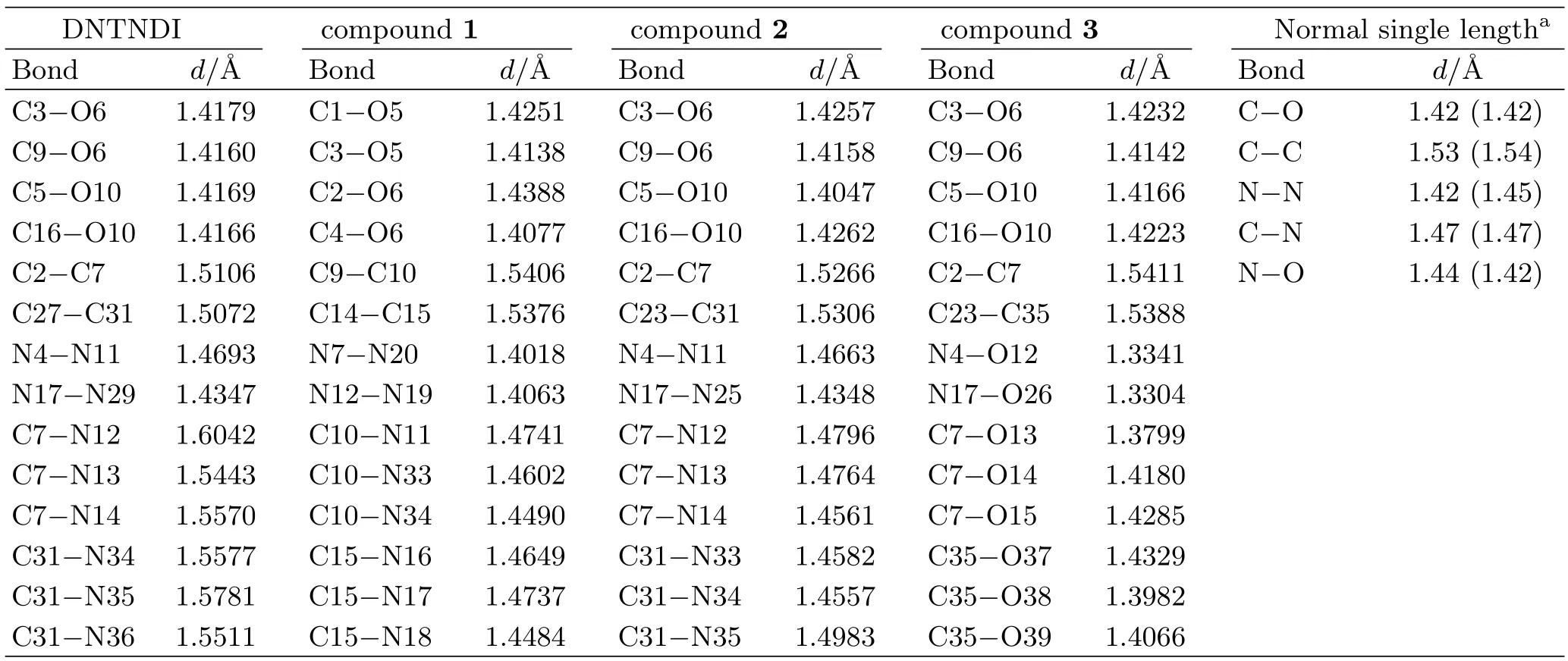
TABLE I Selected bond lengths d(Å)in DNTNDI,and compounds 1,2,3.
It is noted that the C-NO2bond lengths(including C7-N12,13,14 and C31-N34,35,36 single bonds)and N-NO2bond lengths in DNTNDI are much longer than normal single C-N bond and N-N bond,respectively. Besides,the N-NH2bond lengths in compound 1 and the N-ONO2bond lengths in compound 3 are shorter than the bond lengths of N-N and N-O normal single bonds,respectively. The bond lengths of the C-NH2bonds in compound 1 and the C-ONO2bonds in compound 3 are slightly shorter than those of C-N and C-O normal bonds,respectively.
B. Infrared spectra and vibrations
The infrared spectra of DNTNDI and compounds 1,2,3 were predicted using B3LYP/6-311++G∗∗method. Figure 3 presents the calculated infrared spectra.
The infrared vibration modes of these compounds correspond to the peaks. The asymmetric stretching vibrations of O-C-O and C-C-O in the dioxin ring of the title compounds are associated with the peaks of 1156,1129,1164,and 1156 cm-1for DNINDI,and compounds 1,2,3,respectively. There is no split for these peaks,which implies that the vibration coupling effects of the molecular skeletons are not obvious.
Besides,other evident peaks are all associated with the -NO2,-NH2,-N3,and -ONO2groups. It is worth noting that,most of the strongest peaks of these compounds correspond to the asymmetric stretching vibration of the groups(-NO2,-N3,and -ONO2),except compound 1. Compound 1 has the strongest peak at 877 cm-1,which is caused by the wagging vibration of -NH2groups.
The investigations reveal that most of the strong peaks are associated with C-O single bonds in dioxin rings or the bonds between the substituent groups and the imidazole rings. Thereby,the vibrations of these parts in the molecules are easier to be enhanced when the compounds are heated.
C. Heats of formation
HOF is the change of enthalpy from the formation of 1 mole of the compound from its constituent elements,with all substances in their standard states at 1 atmosphere. So in general,the more HOF a compound contains,the more energy is released during the thermal decomposition. From Hesss law,HOF can be used to estimate the amount of energy release in a detonation. Thereby,HOF is a very important factor which determines the detonation properties.
Isodesmic reaction is used to predict the HOFs of DNTNDI,and compounds 1,2,3. For this method,precise HOF data of reference compounds led to accurate prediction[44]. The energies and experimental HOFs of the reference compounds are listed in Table II.
Table III summarizes the total energies,ZPE,thermal corrections,HOFs(in gas and solid phase)of compounds DNTNDI,1,2,3. The results reveal that the DNTNDI and compound 2 exhibit positive HOFs and compound 2 with -N3groups has the largest HOF among all these compounds. The energy of groups can be reflected by the energy of the corresponding methyl compounds. The comparison of experimental HOFs[46]of CH3N3(298.3 kJ/mol),CH3NO2(-80.8 kJ/mol),CH3NH2(-22.5 kJ/mol),and CH3ONO2(-122 kJ/mol)shows that the -N3group has the largest energy among the four groups. Similarly,the -ONO2group has the smallest energy because of the smallest HOF of CH3ONO2.

FIG. 3 The calculated infrared spectra of the title compounds.(a)DNTNDI,(b)compound 1,(c)compound 2,and(d)compound 3.
D. Detonation properties
D and P are key indicators to describe detonation properties for explosives. To estimate the D and P for explosive only containing CHON elements,K-J equations can be applied[36]. Obviously,the crystal density ρ is the most crucial parameter which directly affects D and P. The crystal density of 1.883 g/cm3for DNTNDI was obtained and is slightly smaller than experimental density 1.929 g/cm3[8]. The reason may be that theoretical calculation only involves the single molecule,while the experiment involves the more molecules and considers the interactions among them.
In order to investigate the effect of the different groups on the properties,the molecular surface area A, the electrostatic potential parameters,the oxygen balance OB,the condensed density ρ,and the predicted detonation velocity D and pressure P for the title compounds are summarized and listed in Table IV. The densities and detonation properties of hexahydro-1,3,5-trinitro-1,3,5-triazine(RDX)and 1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane(HMX)are also listed in Table IV for comparison.
The predicted D and P of DNTNDI are 9.15 km/s and 38.14 GPa,respectively,which are close to those of HMX and all larger than those of RDX. The oxygen balance of DNTNDI is zero,which implies that the compound can combust completely,produce the maximum gas products and release the maximum heat. The D(8.15 km/s)and P(30.30 GPa)of compound 3 are smaller than those of RDX. Nevertheless,for compounds 1 and 2,the detonation properties are unsatisfactory. Compounds 1 and 2 have less oxygen and the C atoms cannot be combusted completely,so both the gas phase products and the heat release are reduced. It is observed that imidazole ring doesn't contain O atoms,so the groups containing oxygen atoms,such as -NO2and -ONO2,are necessary to be involved in order to increase the oxygen balance for imidazole derivatives. In addition,it is noted from Table IV that ρ and∆Hf,gasboth play the important role in determining D and P.
E. Frontier molecular orbital and average binding energy analysis
FMO are the electron distributions of the highest occupied energy level(HOMO)and the lowest unoccupied energy level(LUMO),which are associated with the electric and optical properties,as well as the chemical reactivities[51]. The FMO of the title compounds were computed using B3LYP/6-311++G∗∗method and are shown in Fig.4.
The FMOs of all these compounds except compound 1 are localized on the external functional groups,such as -NO2,-N3,and -ONO2groups. Besides,the FMO of compound 1 are localized on the C-N bonds in the imidazole rings. No apparent delocalization effects of electron arise for all the FMOs. The values of the energy gaps between the HOMO and LUMO are 4.49,5.74,4.94,and 4.23 eV for DNINDI and compound 1,2,3,respectively.
The ABEs for DNINDI and compound 1,2,3 were also calculated and are shown in Fig.5. The electronegativities of -NO2,-NH2,-N3,and -ONO2groups are from Ref.[52]. The ABEs,HOMO-LUMO gap,and the reciprocal of the electronegativities for corresponding groups are shown in Fig.4 for comparison. From Fig.5,the ABEs,HOMO-LUMO gaps,and the reciprocal of the electronegativities for the title compounds follow the similar trends. Thus,for the dioxin-imidazole derivatives,the electronegativities of the substituents directly affect the FMO gaps and the ABEs. A larger electronegativity for the substituent leads to a lower HOMO-LUMO gap and a smaller ABE. The FMO gaps and ABEs reveal the higher chemical stability for compound 1 and the larger chemical reactivity for compound 3.
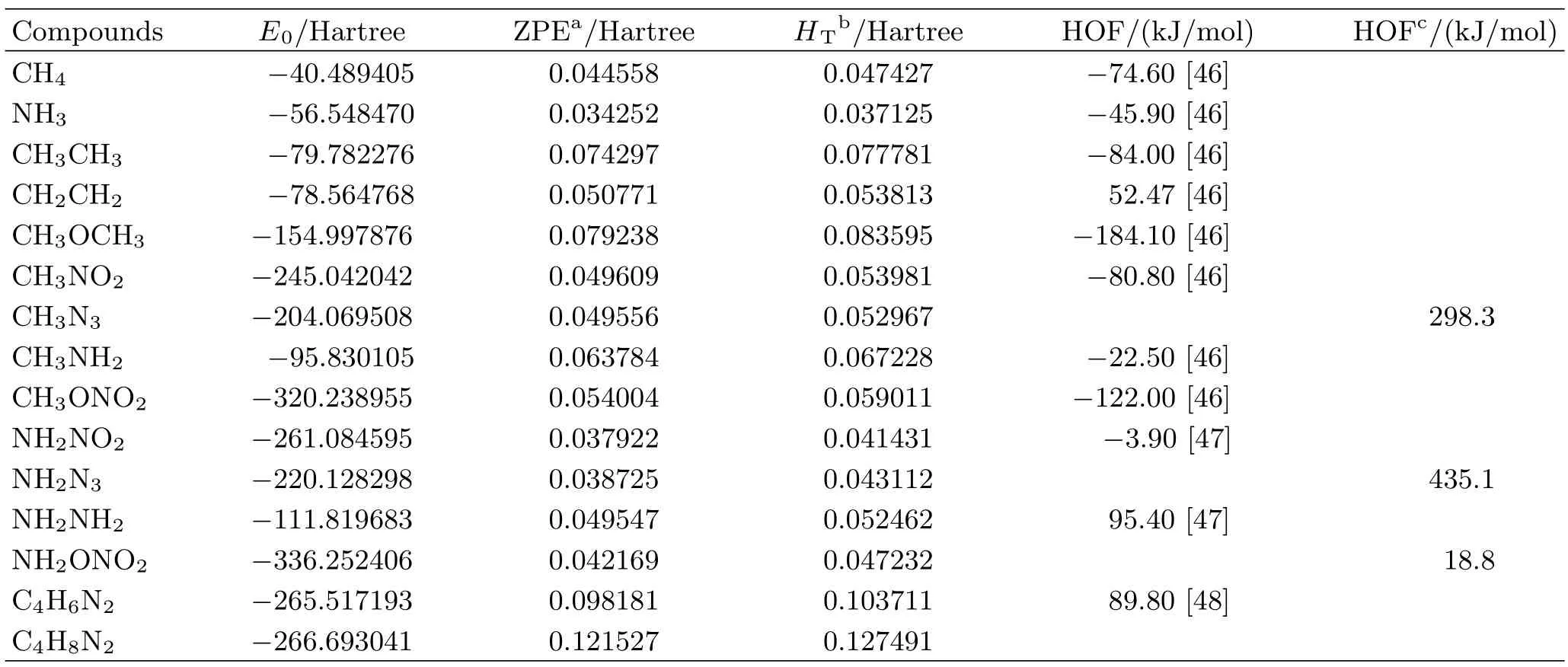
TABLE II Calculated total energies(E0),zero-point energies(ZPE),thermal corrections(HT),and the heats of formation (HOF)of the reference compounds.

TABLE III Calculated total energies(E0),zero-point energies(ZPE),thermal corrections(HT),and HOFs of DNTNDI and compounds 1,2,3.
F. Thermal stability
BDE is often used to describe the strength of bonds,discovers the weakest bond and investigates the pyrolysis mechanism and thermal stability. The NBO analyses of the title compounds were performed to select the weakest bonds. The NBO analysis not only provides the charge transfer interactions between donor NBO and acceptor NBO,but also exhibits the WBI,which gives an indication to the stability of a bond. A smaller WBI value indicates a weaker connection for bonds of same type,while a bigger WBI value implies a stronger linkage.
The bonds of the same type are selected and their WBIs are listed in Table V for comparison. In the Table V,the bonds with the smallest WBI value are considered as the weakest bonds(in bold)for the bondsof same type in each compound and their BDEs are calculated and listed in Table VI.
The electronegativities[52]of -NO2,-N3,and -NH2groups are 3.30,3.04,and 2.42,respectively. Obviously,there are significant inductive effects between the groups and other parts of the molecules. The electron withdrawing groups attract electrons,so the electron populations of their neighbor bonds C-R are reduced. Thus,if a bond is closer to electron withdrawing group or the group is more electronegative,then the bond is weaker. According to the ascending order of electronegativities,the ascending order for BDE of the C-N bonds should be DNTNDI,compound 2,and compound 1,as Table VI shows.
For DNTNDI,compounds 1,and 2,the N-N bonds have the smallest BDE values,but compound 3 locates its weakest bond at the C-O bond in the dioxin ring. The breaking of these bonds may be the first step of thermal decomposition of the compounds. Moreover,the -NH2groups make the compounds the most stable,while the -NO2groups make the compounds the most thermodynamic reactive. Based on data in Table VI,DNTNDI is the least stable compound,while compound 1 is the most stable compound.

TABLE IV Predicted crystal densities ρ,detonation properties D,the electrostatic potential parameters νσ2tot,the oxygen balance OB,the molecular surface area A,and pressure P of the title compounds.

FIG. 4 The calculated FMO and corresponding energy levels of the title compounds based on B3LYP/6-311++G∗∗level.
IV. CONCLUSION
In this work,we investigated the molecular structures,infrared spectroscopy,heats of formation,detonation properties,chemical and thermal stabilities for a series of tetrahydro-[1,4]dioxino[2,3-d:5,6-d′]diimidazole derivatives with four different substituents using hybrid DFT method. Results reveal that the compounds substituted with -NO2and -ONO2groups have larger densities than those with -NH2and -N3groups. The -N3group is a better substituent than the others for increasing the HOF. Most of the strong infrared peaks are associated with the deformation of the substituent groups and the dioxin rings,which indicates that these parts are relatively more reactive areas in the molecules.
The D and P values for the substitution with -NO2group are closer to those of HMX,the D and P values of the derivative with -ONO2group are lower than those of RDX. The investigation for calculated ABEs,HOMO-LUMO gaps and the electronegativities of the substituent groups exhibits that the electronegativity of substituent group directly affects the chemical stability of the compound. For the dioxin-imidazole derivatives,a larger electronegativity for the substituent leads to a lower HOMO-LUMO gap and a smaller ABE. The cal-culations for BDEs indicate that the N-N bonds are the weakest bonds for DNTNDI and compounds 1,2. The C-O bonds in the dioxin rings have the smallest BDE for the compound 3. Considering the detonation properties and thermal stabilities,the DNTNDI can be candidate for high energy density materials.

FIG. 5 The calculated ABEs and HOMO-LUMO gaps for DNTNDI and compounds 1,2,3,as well as reciprocal of the electronegativities for the corresponding substituents[52].

TABLE V The Wiberg bond index of the title compounds from NBO analysis.

TABLE VI The calculated BDEs of the selected bonds of the title compounds.
V. ACKNOWLEDGMENTS
This work was supported the National Natural Science Foundation of China(No.U1304111),and the Program for Science & Technology Innovation Talents in Universities of Henan Province(No.14HASTIT039)and the Innovation Team of Henan University of Science and Technology(No.2015XTD001).
[1]S. Bulusu,R. Damavarapu,and J. R. Autera,J. Phys. Chem. 99,5009(1995).
[2]S. G. Cho,Y. G. Cheun,and B. S. Park,J. Mol. Struct. 432,41(1998).
[3]P. R. Singh,H. X. Gao,and D. T. Meshri,Struct. Bond. 125,35(2007).
[4]D. M. Badgujar,M. B. Talawar,and S. N. Asthana,J. Hazard Mater. 151,289(2008).
[5]A. T. Nielsen,A. P. Chafin,S. L. Christian,D. W. Moore,M. P. Nadler,R. A. Nissan,D. J. Vanderah,R. D. Gilardi,C. F. George,and J. L. Flippen-Anderson,Tetrahedron 54,11793(1998).
[6]M. Vedachalam,V. T. Ramakrishnan,J. H. Boyer,I. J. Dagley,K. A. Nelson,H. G. Adolph,R. Gilardi,C. George,and J. L. Flippen-Anderson,J. Org. Chem. 56,3413(1991).
[7]H. Rongzu,L. Xingsen,and F. Yingao,J. Energetic Mater. 11,219(1993).
[8]M. J. Wu,S. Chen,Q. H. Shu,L. J. Li,and S. H. Jin,Propellants Explos. Pyrotech. 38,658(2013).
[9]Y. L. Sun and S. F. Li,J. Hazard. Mater. 154,112 (2008).
[10]S. Dharavath,D. G. Vikas,P. T. Surya,and M. Krishnamurthi,Chem. A Eur. J 47,18(2012).
[11]N. S. Karanjule,M. B. Talawar,R. Sivabalan,G. M. Gore,D. D. Dhavale,and S. N. Asthana,Indian J. Chem. Technol. 14,34(2007).
[12]X. W. Fan and X. H. Ju,J. Comput. Chem. 29,505 (2008).
[13]B. M. Rice,A. V. Pai,and J. Hare,Combust Flame 118,445(1999).
[14]X. H. Li,R. Z. Zhang,and X. Z. Zhang,J. Hazard Mater. 183,622(2010).
[15]F. J. Owens,J. Mol. Struct. 370,11(1996).
[16]C. F. Melius and S. N. Bulusu,Chemistry and Physics of Energetic Materials,The Netherlands:Kluwer Academic Publishers,158(1990).
[17]B. M. Rice,S. Sahu,and F. J. Owens,J. Mol. Struct. 583,69(2002).
[18]S. W. Zhang and T. N. Truong,J. Phys. Chem. A 104,7304(2000).
[19]A. D. Becke,J. Chem. Phys. 98,5648(1993).
[20]C. Lee,W. Yang,and R. G. Parr,Phys. Rev. B 37,785 (1988).
[21]B. D. Becke,Phys. Rev. B 38,3098(1988).
[22]P. J. Stephens,F. J. Devlin,C. F. Chabalowski,and M. J. Frisch,J. Phys. Chem. 98,11623(1994).
[23]L. Qiu,H. M. Xiao,X. D. Gong,X. H. Ju,and W. H. Zhu,J. Hazard. Mater. 141,280(2007).
[24]M. J. Frisch,G. W. Trucks,H. B. Schlegel,G. E. Scuseria,M. A. Robb,J. R. Cheeseman,J. A. Jr. Montgomery,T. Vreven,K. N. Kudin,J. C. Burant,J. M. Millam,S. S. Iyengar,J. Tomasi,V. Barone,B. Mennucci,M. Cossi,G. Scalmani,N. Rega,G. A. Petersson,H. Nakatsuji,M. Hada,M. Ehara,K. Toyota,R. Fukuda,J. Hasegawa,M. Ishida,T. Nakajima,Y. Honda,O. Kitao,H. Nakai,M. Klene,X. Li,J. E. Knox,H. P. Hratchian,J. B. Cross,C. Adamo,J. Jaramillo,R. Gomperts,R. E. Stratmann,O. Yazyev,A. J. Austin,R. Cammi,C. Pomelli,J. W. Ochterski,P. Y. Ayala,K. Morokuma,G. A. Voth,P. Salvador,J. J. Dannenberg,V. G. Zakrzewski,S. Dapprich,A. D. Daniels,M. C. Strain,¨O. Farkas,D. K. Malick,A. D. Rabuck,K. Raghavachari,J. B. Foresman,J. V. Ortiz,Q. Cui,A. G. Baboul,S. Clifford,J. Cioslowski,B. B. Stefanov,G. Liu,A. Liashenko,P. Piskorz,I. Komaromi,R. L. Martin,D. J. Fox,T. Keith,M. A. Al-Laham,C. Y. Peng,A. Nanayakkara,M. Challacombe,P. M. W. Gill,B. Johnson,W. Chen,M. W. Wong,C. Gonzalez,and J. A. Pople,Gaussian 03,Revision C. 01,Wallingford,CT:Gaussian,Inc.(2003).
[25]J. B. Foresman and A. Frisch,Exploring Chemistry with Electronic Structure Methods,2nd Edn.,Pittsburg:Gaussian,Inc.(1996).
[26]S. Y. Lee and B. H. Boo,Bull. Korean Chem. Soc. 17,754(1996).
[27]P. L. Fast,J. Corchado,H. L. Sanchez,and D. G. Truhlar,J. Phys. Chem. A 103,3139(1999).
[28]M. Kurt and S. Yurdakul,J. Mol. Struct. 654,1(2003).
[29]M. Selmi and J. Tomasi,J. Phys. Chem. 99,5894 (1995).
[30]Z. X. Chen,J. M. Xiao,H. M. Xiao,and Y. N. Chiu,J. Phys. Chem. A 103,8062(1999).
[31]H. M. Xiao and Z. X. Chen,The Modern Theory For Tetrazole Chemistry,1st Edn.,Beijing:Science Press,(2000).
[32]P. C. Chen,Y. C. Chieh,and S. C. Tzeng,J. Mol. Struct. 634,215(2003).
[33]P. Politzer and J. S. Murray,Energetic Materials,Amsterdam:Elsevier,(2003).
[34]P. Politzer,J. S. Murray,M. E. Grice,M. DeSalvo,and E. Miller,Mol. Phys. 91,923(1997).
[35]E. F. C. Byrd and B. M. Rice,J. Phys. Chem. A 110,1005(2006).
[36]M. J. Kamlet and S. J. Jacobs,J. Chem. Phys. 48,23 (1968).
[37]C. Hollister and O. Sinanoglu,J. Am. Chem. 88,1 (1966).
[38]A. E. Reed,L. A. Curtiss,and F. Weinhold,Chem. Rev. 88,899(1988).
[39]S. W. Benson,Thermochemical Kinetics,2nd Edn.,New York:Wiley-Interscience,176(1976).
[40]X. Q. Yao,X. J. Hou,G. S. Wu,Y. Y. Xu,H. W. Xiang,H. Jiao,and Y. W. Li,J. Phys. Chem. A 106,7184(2002).
[41]J. Shao,X. Cheng,and X. Yang,J. Mol. Struc-Theochem. 127,755(2005).
[42]X. W. Fan,X. H. Ju,Q. Y. Xia,and H. M. Xiao,J. Hazard. Mater. 151,255(2008).
[43]Y. Sasada,Molecular and Crystal Structures in Chemistry Handbook,Tokyo:The Chemical Society of Japan,119(1984).
[44]X. H. Li,Y. L. Yong,and X. Z. Zhang,Mol. Phys. 112,1040(2014).
[45]A. P. Scott and L. Radom,J. Phys. Chem. 100,16502 (1996).
[46]J. A. Dean,Lange′s Handbook of Chemistry,15th Edn.,Knoxville TN. USA:McGRAW-HILL,INC. Sec. 6. 5-50(1999)
[47]R. L. David,Handbook of Chemistry and Physics,84th Edn.,Boca Raton:CRC Press,1038(2004).
[48]P. Jimenez,M. V. Roux,and C. Turrion,J. Chem. Thermodyn. 24,1145(1992).
[49]M. B. Talawar,R. Sivabalan,T. Mukundan,H. Muthurajan,A. K. Sikder,B. R. Gandhe,and A. S. Rao,J. Hazard. Mater. 161,589(2009).
[50]D. M. Badgujar,M. B. Talawar,S. N. Asthana,and P. P. Mahulikar,J. Hazard. Mater. 151,289(2008).
[51]J. Fleming,Frontier Orbitals and Organic Chemical Reactions,London:Wiley,(1976).
[52]S. G. Bratsch,J. Chem. Edu. 62,101(1985).
DOI:10.1063/1674-0068/29/cjcp1506118
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Impact of Eu3+Ions on Physical and Optical Properties of Li2O-Na2O-B2O3Glass
- Investigation of Ultrafast Electronic Transfer Process on Organic/Inorganic Heterojunction by Femtosecond Transient Absorption
- Ordered Toroid Structures of Nanoparticles in Self-attractive Semiflexible Polymer/Nanoparticle Composites
- Virtual Screening of Human O-GlcNAc Transferase Inhibitors
- Morphology and Growth Process of Bat-like ZnO Crystals by Thermal Evaporation
- Epitaxial Growth and Thermoelectric Measurement of Bi2Te3/Sb Superlattice Nanowires