Virtual Screening of Human O-GlcNAc Transferase Inhibitors
2016-07-05QingtongZhouHaojunLiangEugeneShakhnovichaDepartmentofPolymerScienceandEngineeringCASKeyLaboratoryofSoftMatterChemistryUniversityofScienceandTechnologyofChinaHefei230026ChinaDepartmentofChemistryandChemicalBiologyHarvar
Qing-tong Zhou,Hao-jun Liang,Eugene Shakhnovicha. Department of Polymer Science and Engineering,CAS Key Laboratory of Soft Matter Chemistry,University of Science and Technology of China,Hefei 230026,China b. Department of Chemistry and Chemical Biology,Harvard University,Cambridge MA 02138,USA
Virtual Screening of Human O-GlcNAc Transferase Inhibitors
Qing-tong Zhoua,b,Hao-jun Lianga∗,Eugene Shakhnovichb∗
a. Department of Polymer Science and Engineering,CAS Key Laboratory of Soft Matter Chemistry,University of Science and Technology of China,Hefei 230026,China b. Department of Chemistry and Chemical Biology,Harvard University,Cambridge MA 02138,USA
(Dated:Received on October 14,2015;Accepted on December 10,2015)
O-GlcNAc transferase(OGT)is one of essential mammalian enzymes,which catalyze the transfer of N-acetylglucosamine from UDP-N-acetylglucosamine(UDP-GlcNAc)to hydroxyl groups of serines and threonines(Ser/Thr)in proteins. Dysregulations of cellular O-GlcNAc have been implicated in diabetes,neurodegenerative disease,and cancer,which brings great interest in developing potent and specific small-molecular OGT inhibitors. In this work,we performed virtual screening on OGT catalytic site to identify potential inhibitors. 7134792 drug-like compounds from ZINC(a free database of commercially available compounds for virtual screening)and 4287550 compounds generated by FOG(fragment optimized growth program)were screened and the top 116 compounds ranked by docking score were analyzed. By comparing the screening results,we found FOG program can generate more compounds with better docking scores than ZINC. The top ZINC compounds ranked by docking score were grouped into two classes,which held the binding positions of UDP and GlcNAc of UDPGlcNAc. Combined with individual fragments in binding pocket,de novo compounds were designed and proved to have better docking score. The screened and designed compounds may become a starting point for developing new drugs.
Key words:O-GlcNAc transferase,Virtual screening,Inhibitor,ZINC,FOG,Drug design
∗Authors to whom correspondence should be addressed. E-mail:hjliang@ustc.edu.cn,shakhnovich@chemistry.harvard.edu,Tel./ FAX:+86-551-63607824
I. INTRODUCTION
O-linked N-acetylglucosamine cylation(OGlcNAcylation)[1]is one kind of protein posttranslational modification. Due to its important effect on protein stability,subcellular localization,phosphorylation and ubiquitination,the dysregulation of cellular O-GlcNAc has been implicated in diabetes,neurodegenerative disease and cancer. There are two enzymes responsible for the regulation of O-GlcNAc:one is O-GlcNAc transferase(OGT),the other is OGlcNAc hydrolase(OGA)which cleaves the glycosidic bond that reverses the modification. OGT is comprised by a multidomain catalytic region,an N-terminal region with different numbers of tetratricopeptide repeats(TPRs)and a linker region. TPRs are far from the catalytic site and not essential for glycosyl transfer onto acceptor peptides,while it is thought to be closely related to OGT subcellular localizations. There are at least three different isoforms of human OGT:nucleocytoplasmic OGT(ncOGT)contains 12.5 TPRs,mitochondrial OGT(mOGT)contains 9.5 TPRs and short OGT(sOGT)2.5 TPRs. Given the importance of O-GlcNAc modifications in signaling pathways,several potent and selective human O-GlcNAc transferase (hOGT)inhibitors were designed. Alloxan is the first reported hOGT inhibitor[2],which is thought to inhibit hOGT by binding to the uracil binding pocket or alternatively has been proposed to act through a covalent modification of cysteine residues. Besides,benzyl 2-acetamido-2-deoxy-α-D-galactopyranoside (BAGDP)is reported to perturb O-GlcNAc in cells,while it is not clear whether it could selectively inhibit OGT in cells. Several crystal structure of OGT bound with UDP-GlcNAc analogs[3-6]including UDP-5SGlcNAc,UDP-S-GlcNAc,UDP-GlcNAcF3,SP-αS-UDP-GlcNAc,RP-αS-UDP-GlcNAc were determined to reveal the mechanisms of GlcNAc transfer and substrate recognition. Experimental high-throughput screening(HTS)[7-9]has been greatly involved in hOGT inhibitors discovery and identified several novel hOGT inhibitors,while the challenge of building robust nonradiometric assay strategies to detect glycosylation limits its application.
With the development of computational chemistry and biology[10-13],virtual screening[14-19]is becoming increasing important in identifying novel compounds in many drug discovery projects. Recently,several computational techniques like molecular dynamics simulation[20,21],molecular docking[22-24],QSAR [25],pharmacophore designing[25-27]have been developed to speed up the drug discovery process. Asthe chemical space of all possible chemical structures is extraordinarily large,it is reasonable to screen a drug target against a selection of chemicals which cover as much of the appropriate chemical space as possible. To address that,chemical libraries can be classified as diverse oriented,drug-like,natural product-like,and targeted against a specific family of biological targets such Kinases,GPCRs,PPI etc. ZINC[28](drugs now)is one widely-used database containing only compounds with drug-like properties(“Lipinski's rule of five”)[29]. FOG library is prepared by fragment optimized growth (FOG)program[30]in Shakhnovich group,which statistically biases the growth of molecules with desired features,such as stability in water,synthetic accessibility,or druglikeness[31]. Here FOG(FDA approved drugs)was prepared by generating new compounds in a chemical space that is similar to the compounds that were trained on 2713 drugs from Hutter database,which has better druglikeness than with a random growth.
The present study was to search for potential inhibitors for OGT started from FOG and ZINC compound database. We chose one of the best molecular docking programs available currently GLIDE[32]and performed virtual screening to identify the potential inhibitors. We investigated the practical application of FOG and found FOG library contains more compounds with better docking scores than ZINC. By the combination of individual fragments in binding pocket,we designed new compounds with better docking score,which could be a start-point for lead optimizations.
II. COMPUTATIONAL DETAILS
A. Protein and grid preparation
The crystal structure of hOGT with UDP-GlcNAc (PDB ID:4GZ5)[3,33]determined a resolution of 3.08Å was retrieved from the Protein Data Bank. We adopted the hOGT with UDP(PDB ID:3PE3)and UDP-5SGlcNAc(PDB ID:4GZ6)complexes as references. The structures were prepared using the protein preparation wizard of Schr¨odinger module. OPLS-2005 force field was used for energy minimization to acquire an energetically stable geometry. Hydrogen atoms were added to the protein to correct ionization and tautomeric states of amino acid residues. The shape and properties of the receptors were represented on a grid by several different sets of fields that provide progressively accurate scoring of the ligand poses using Receptor Grid Generation Panel. We have generated the grid that covers all the catalytic residues with peptide in the cavity.
B. Ligand library preparation
The ligand library including 7134792 drug-like compounds was extracted from the ZINC database (http://zinc.docking.org/subsets/drugs-now). FOG library is composed by 4287550 compounds generated by FOG program. These compounds were prepared in Lig-Prep Wizard where hydrogens were added and followed by minimization and optimization in OPLS-2005 force field,while the original chirality were retained.
C. Virtual screening study
Virtual Screening Workflow in Maestro was used to dock the lead-like compounds to identify potential ligands. The Glide(grid-based ligand docking with energetic)algorithm approximates a systematic search of positions,orientations,and conformations of the ligand in the receptor binding site using a series of hierarchical filters. There are three different level of docking precision,namely high throughput virtual screening (HTVS),standard precision(SP)and extra precision (XP). The XP glide scoring function[32]is presented in Eq.(1)while the favorable binding terms and unfavorable terms are presented in Eq.(2)and Eq.(3),respectively.

The contributions from the Coulomb and van der Waals protein-ligand interaction energies by Schr¨odinger's “active-site mapping”technology are Ecouland EvdW. Ehb-pairis standard ChemScore-like hydrogen bond term while Ephobic-pairis lipophilic pair term. To improve the accuracy of favorable binding terms Ebind,hydrophobic interactions enclosure Ehyd-enclosure,special neutral-neutral hydrogen-bond motifs Enb-nn-motif,special charged-charged hydrogen-bond motifs Ehb-cc-motif,and π stacking and π-cation interactions rewarded terms EPIwere introduced to the score function. Strain energy of the ligand and/or protein,loss of entropy of ligand and protein,and desolvation of the ligand or protein are adverse to the binding,thus Edesolv(rapid docking of explicit waters)and Eligand−strain(contact penalties)were developed to penalize XP binding score. Compared with other docking programs[11],Glide has the advantage on finding the correct binding modes for a large set of test cases,especially the predicted binding poses have lower RMS deviations from native co-crystallized structures. As a complete solution for ligand-receptor docking,Glide is continuously optimized to improve speed and accuracy. Thus in the current work,Glide was adopted to perform ligandreceptor docking.
All compounds was first screened on chain D of 4GZ5 through HTVS stage,then the top 8%from ZINC and the top 15%from FOG were subjected to SP stage. After SP docking,the top 5%from ZINC and the top 6%from FOG were subjected to XP docking(Table I). Finally,we selected the top 116 ranked by docking score compounds and performed multiple receptor conformations docking to model protein flexibility[34],i.e.,we docked them to other 11 OGT receptors(chain A,B,C of 4GZ5,chain A,B,C,D of 4GZ6 and 3PE3)to obtain averaged docking score.
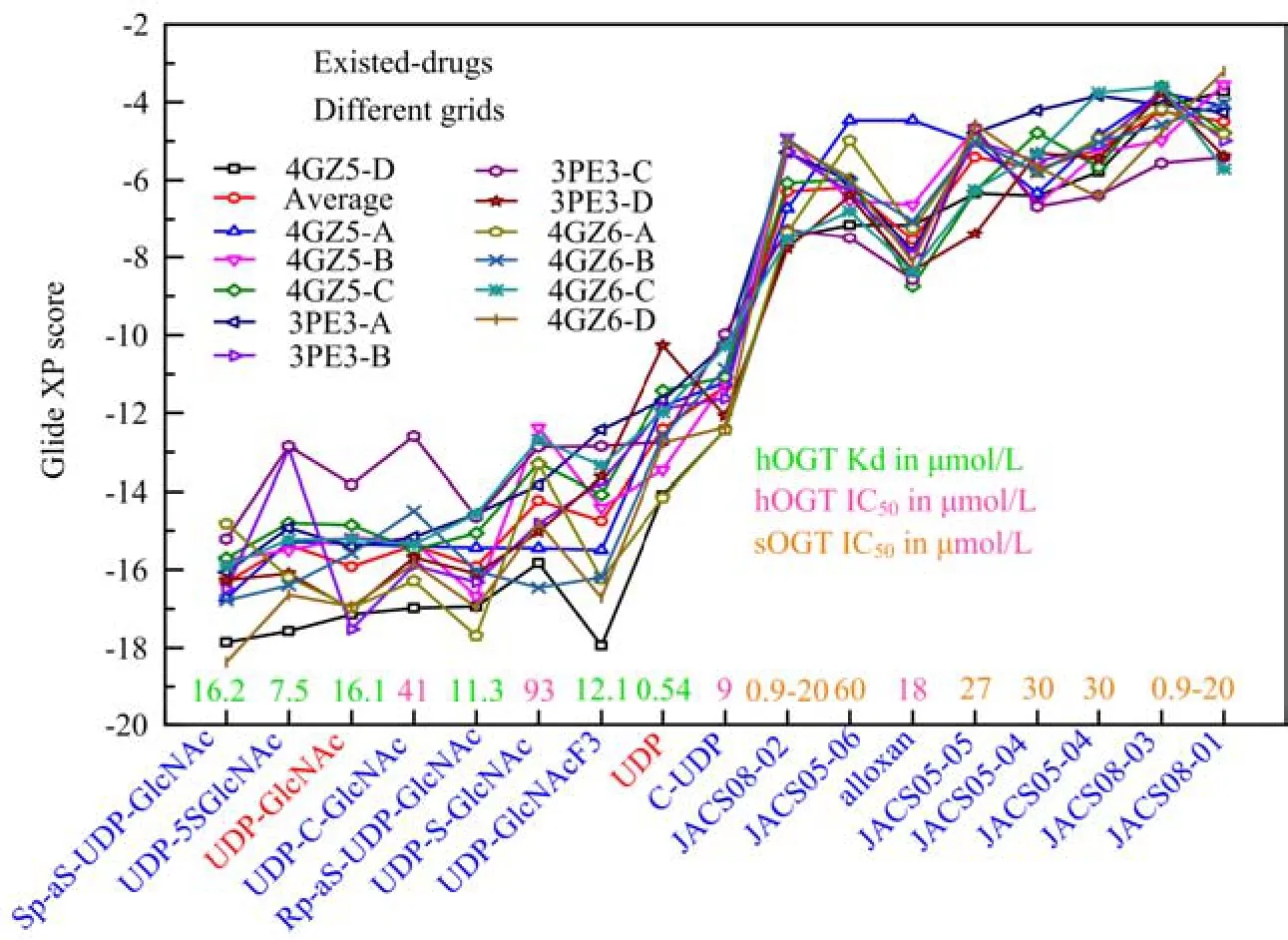
FIG. 1 Docking score of UDP-GlcNAc analogs and experimental discovered inhibitors by Glide XP.
III. RESULTS AND DISCUSSION
A. Virtual screening
UDP-GlcNAc analogs and several experimental OGT inhibitors[7,8,35]were docked to OGT to evaluate the performance of docking program Glide. As presented in Fig.1,there were obvious difference in docking score between UDP-GlcNAc analogs and experimental discovered inhibitors. We found the predicted binding conformations of UDP-GlcNAc analogs are in consistence with experimental crystal structures,the averaged docking score of UDP and UDP-GlcNAc on twelve receptors (chain A,B,C,and D of 4GZ5,4GZ6 and 3PE3)are -12.40(the more negative,the better)and -15.91,respectively. The first reported hOGT inhibitor alloxan (IC50=18±1µmol/L)has an averaged docking score -7.56(the lowest is -8.74,chain C of 4GZ5),which supports the thought that alloxan is an efficient binder in terms of ligand efficiency[35]. Three validated OGT inhibitors discovered through HTS[8]were docked to OGT with docking score ranging from -4.38 to -6.30,which agrees with their IC50values between 0.9 and 20µmol/L. In current research,we chose UDP,the docking score is -12.40,as one reference.

TABLE I Screened ligand library in virtual screening study. Subset of FOG are named according to molecular weight (MW),like MW-250 is comprised by the compounds whose molecular weight ranged from 250 g/mol to 300 g/mol.
The top ten FOG and ZINC compounds are listed in Table II. The averaged docking score of FOG have ranges of -10.41 to -11.17,while that of ZINC is -9.75 to -11.20. The lowest docking score on chain D of 4GZ5 are -12.97 and -13.39 for FOG and ZINC,respectively. As presented in Fig.2,we found,sorted by the averaged docking score,FOG contain more compounds with lower docking score than ZINC,which demonstrated the advantage of FOG in library preparation during virtual screening. Unfortunately,the best ZINC and FOG compounds isolated by HTVS have higher docking score than UDP,which means these compounds can hardly compete with UDP(IC50=1.8±1.0µmol/L)inthe binding site. To address this question,we performed virtual screening based drug design.
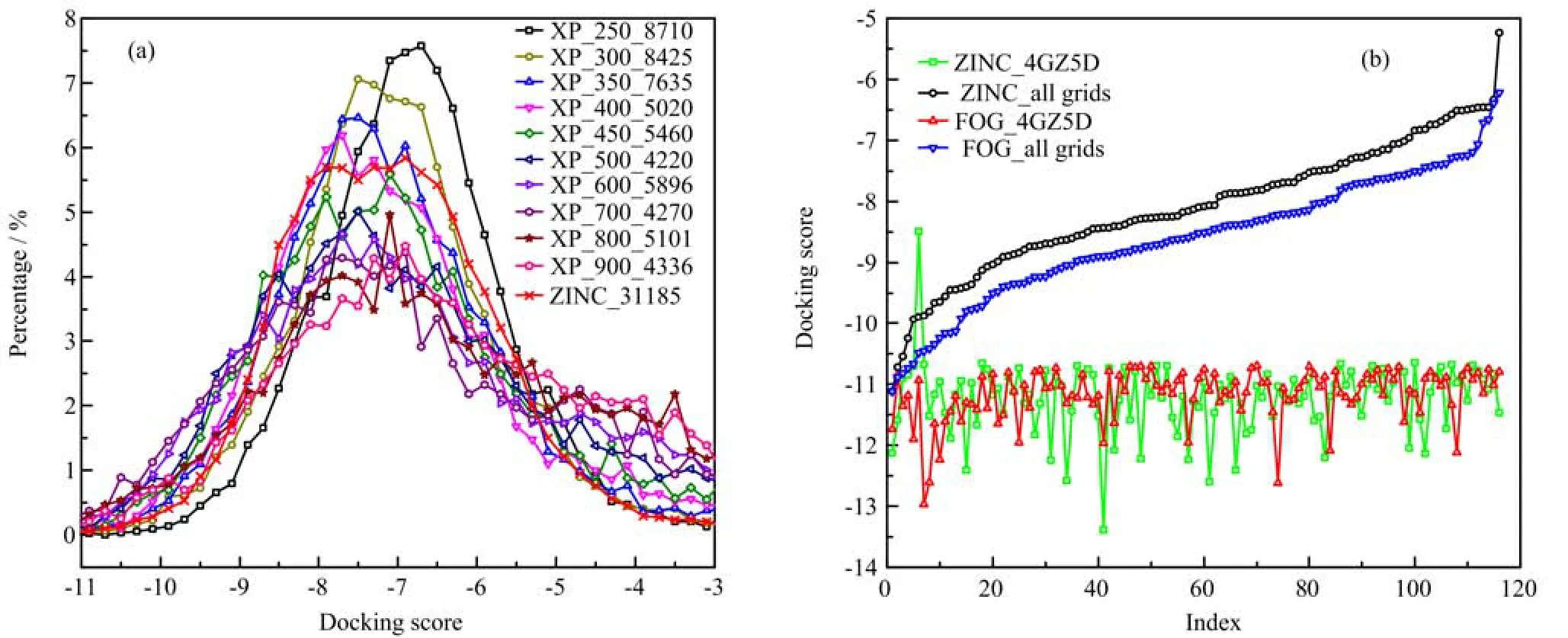
FIG. 2 (a)Distribution of FOG and ZINC library over the values of docking score at XP stage. MW-250 library is generated by FOG program and comprised by the compounds whose molecular weight ranged from 250 g/mol to 300 g/mol. (b)Comparison of the averaged docking score for the top 116 FOG and ZINC compounds.

TABLE II Docking score of the top ten compounds from FOG and ZINC Library. The averaged docking scores of the twelve receptors(chain A,B,C and D of 4GZ5,4GZ6 and 3PE3)were determined,listed as“twelve grids”. FOG compounds whose molecular weight are lower than 500 g/mol are chosen to compare with ZINC library.
B. FOG and ZINC
FOG program generated new compounds by adopting the parameters which were trained on Hotter database (2713 drugs),thus the produced compounds enjoy the desired features such as druglikeness,stability in water,while there's not clear in its performance during virtual screening. To evaluate practical value of FOG in drug screening,we compared it with ZINC at two aspects:docking score distribution and fragment novelty of chemical compounds.
At HTVS stage,only 74.4%compounds from FOG can be docked to OGT while that value for ZINC is 95.3%. At SP stage,we found as the MW of FOG compounds increasing,the distribution of docking score move to the left,that means the compounds with higher MW tend to have lower docking scores. At XP stage,ZINC compounds concentrated in the range from -6 to -8.5,while FOG is more widely distributed. 1.04% of XP-screening ZINC compounds have lower docking score than -10. As MW of FOG compound increased,the percentage of compounds that located at low score region(docking score<-10)grow rapidly,from 0.21% for FOG MW-250 subset,1.50%for FOG MW-400 subset,to 3.29%for FOG MW-600 subset. These results illustrated at higher precision of XP,the FOG provides more compounds with lower docking score.
As presented in Fig.3,the top three ZINC compounds (chain D of 4GZ5)shared 2-hydroxybenzimidazole group,which form hydrogen bonds with Ala896,ππ stacking interaction with Phe868,His901. Among 116 best ZINC compounds,the functional group 2-hydroxybenzimidazole were observed sixteen times,and most of them occupied identical position in the binding pocket. As for the best three FOG compounds,there are distinctly structural diverse but could strongly interact with the residues around the binding pockets,which bring the low docking score. We performed structure-based clustering in a hierarchical manner for the top 116 FOG and ZINC compounds using JKlustor [36]. 116 best ZINC compounds were clustered into 8 top level cluster count and 45 top cluster count while 116 best FOG compounds were clustered into 15 toplevel cluster count and 112 top cluster count. Thus FOG program have generated more novel fragments when compared with ZINC,which would be of great importance for further drug design.

FIG. 3 Chemical structures of three OGT inhibitors with the highest docking scores from ZINC and FOG virtual screening. Docking score is based on the receptor chain D of 4GZ5.
C. Drug design
Although screened ZINC(drugs now)library has drug-like properties(“Lipinski's rule of five”)that is expected to have satisfactory physicochemical properties as drug candidates,the best compounds like ZINC-32,ZINC-11,ZINC-14 have about 2-3 higher docking score than that of UDP,that means they are poor lead compounds for drug discovery because of the high IC50. Therefore,we decided to make chemical modifications on the identified inhibitors to design more potent OGT inhibitors with at least UDP-level inhibitory activity.
The best docked compounds from ZINC were grouped into two classes which held the binding positions of UDP and GlcNAc of UDP-GlcNAc. Compounds like ZINC-11,ZINC-22,ZINC-80,and ZINC-89 formed hydrogens bonds with Thr560,Leu653,His920,Thr921,Thr922 of OGT in the same positions of the GlcNAc part of UDP-GlcNAc,which formed hydrogens bonds with Thr560,Leu653,His498,Gly654,His920 and Thr921. The docked conformations of other compounds have significant overlap with the UDP of UDP-GlcNAc. Take ZINC-14 for an example,2-hydroxybenzimidazole group form hydrogen bonds with Ala896,π-π stacking interaction with Phe868,His901,while nucleotidase uracil of UDP form hydrogen bond with Ala896 and is surrounded by Phe868,His901. The pyrophosphate group and the pentose sugar ribose of UDP contribute nine hydrogens bonds with Lys842,Lys898,His920,Thr921,Thr922,Asp925. Limited by the number of hydrogen bonds donors and acceptors,ZINC-14 form five hydrogen bonds with Gln839,Thr921,His920,and Thr922. The different orientation of potential ZINC inhibitors distributed in the binding pocket brings the probability of linking individual fragments to design new inhibitors.
Following compound combination strategy,we designed a new compound D230-211(Fig.4),which is the combination of ZINC-11 and ZINC-14. As shown in Table III,the averaged docking scores of ZINC-11 and ZINC-14 are -11.20 and -9.65,respectively. The de novo designed compound D230-211 has much better docking score,-12.22,which is very close to UDP. That means the designed compound can inhibit OGT catalysis activity similar to UDP. But compared with UDPGlcNAc's docking score(-15.91),the designed compound is far from full inhibition of OGT activity,that means the dose could be reasonable high. Nonetheless,current design strategy can be applied for general drug design projects.
IV. CONCLUSION
O-GlcNAc transferase(OGT)is one of essential enzymes in protein glycosylation regulations. In this work,we performed virtual screening on ZINC(drugs now)and FOG compound library to identify OGT inhibitors. Through the XP docking on UDP-GlcNAc analogs,we verified that Glide can correctly predict the binding conformations with very low docking score. We found that all of ZINC and FOG compounds have higher docking score than UDP and UDP-GlcNAc,which suggest the potential inhibitors do not naturally exist in the library. Following compound combination strategy,compound D230-211,whose docking score is close to UDP,were designed and could be used as a start point for further lead optimization. We investigated the practical value of FOG in drug screening from the aspects:docking score distribution and novelty of chemical compounds. We found FOG library generate more compounds with better docking scores and present remarkable structure diversity simultaneously,which will be of great practical significance to drug screening.

FIG. 4 Binding poses of two ZINC compounds(ZINC-14 and ZINC-11),which occupied two different regions of the OGT catalytic site. As the combination of two compounds,de novo compounds D230-211 was designed and proved to have better docking score,which suggest a good strategy in further drug design.

TABLE III Docking score of de novo compounds designed by compound combination strategy.
V. ACKNOWLEDGMENTS
This work is supported by the FAS Division of Science,Research Computing Group at Harvard University and the Supercomputing Center of USTC by the National Natural Science Foundation of China (No.20934004 and No.91127046),the National Institutes of Health(No.GM068670),and the China Scholarship Council.
[1]Y. Wang,Acta Chim. Sin. 71,1477(2013).
[2]R. J. Konrad,F. Zhang,J. E. Hale,M. D. Knierman,G. W. Becker,and J. E. Kudlow,Biochem. Biophys. Res. Commun. 293,207(2002).
[3]M. B. Lazarus,J. Jiang,T. M. Gloster,W. F. Zandberg,G. E. Whitworth,D. J. Vocadlo,and S. Walker,Nat. Chem. Biol. 8,966(2012).
[4]J. Jiang,M. B. Lazarus,L. Pasquina,P. Sliz,and S. Walker,Nat. Chem. Biol. 8,72(2012).
[5]M. Schimpl,X. Zheng,V. S. Borodkin,D. E. Blair,A. T. Ferenbach,A. W. Schuttelkopf,I. Navratilova,T. Aristotelous,O. Albarbarawi,D. A. Robinson,M. A. Macnaughtan,and D. M. van Aalten,Nat. Chem. Biol. 8,969(2012).
[6]I. Tvaroska,S. Kozmon,M. Wimmerova,and J. Koca,J. Am. Chem. Soc. 134,15563(2012).
[7]B. J. Gross,B. C. Kraybill,and S. Walker,J. Am. Chem. Soc. 127,14588(2005).
[8]B. J. Gross,J. G. Swoboda,and S. Walker,J. Am. Chem. Soc. 130,440(2008).
[9]R. F. Ortiz-Meoz,J. Jiang,M. B. Lazarus,M. Orman,J. Janetzko,C. Fan,D. Y. Duveau,Z. W. Tan,C. J. Thomas,and S. Walker,ACS Chem. Biol. 10,1392 (2015).
[10]K. Y. Yang,B. Yang,and Z. J. Lin,Chin. J. Chem. Phys. 28,161(2015).
[11]T. J. Cheng,X. Li,Y. Li,Z. H. Liu,and R. X. Wang,J. Chem. Inf. Model. 49,1079(2009).
[12]W. Deng,B. Zheng,W. Ding,H. Zhu,and H. J. Liang,Chin. J. Chem. Phys. 28,630(2015).
[13]H. G. Xu and H. J. Liang,Chin. J. Chem. Phys. 27,679(2014).
[14]T. J. Hou and X. J. Xu,Curr. Pharm. Design 10,1011 (2004).
[15]S. Sirois,D. Q. Wei,Q. S. Du,and K. C. Chou,J. Chem. Inf. Comput. Sci. 44,1111(2004).
[16]Y. Xu,Y. L. Cu,Q. C. Zheng,H. X. Zhang,and C. C. Sun,Chem. J. Chin. U 34,1226(2013).
[17]W. Song,W. Z. Chen,X. Y. Zhang,and C. X. Wang,Chin. J. Chem. Phys. 16,257(2003).
[18]Y. Y. Hao,C. H. and D. Yang,J. Beijing Normal Unviersity(Natural Science)49,6(2013).
[19]F. Bai,Y. Xu,J. Chen,Q. Liu,J. Gu,X. Wang,J. Ma,H. Li,J. N. Onuchic,and H. Jiang,Proc. National Acad. Sci. USA 110,4273(2013).
[20]H. Zhu,S. Xiao,and H. Liang,PloS ONE 8,e71380 (2013).
[21]K. Wang,Y. F. Zhao,G. W. Lu,Y. L. Wang,J. N. Chen,and D. Z. Su,Chin. J. Chem. Phys. 27,380 (2014).
[22]G. Xia,M. Xue,L. Liu,J. Yu,H. Liu,P. Li,J. Wang,Y. Li,B. Xiong,and J. Shen,Bioorg. Med. Chem. Lett. 21,5739(2011).
[23]I. Nagpal,I. Raj,N. Subbarao,and S. Gourinath,PloS One 7,e30305(2012).
[24]B. Xu,Y. Yang,H. Liang,and Y. Zhou,Proteins 76,718(2009).
[25]X. H. Li,R. Z. Zhang,X. L. Cheng,and X. D. Yang,Chin. J. Chem. Phys. 20,167(2007).
[26]X. Liu,S. Ouyang,B. Yu,Y. Liu,K. Huang,J. Gong, S. Zheng,Z. Li,H. Li,and H. Jiang,Nucleic Acids Res. 38,W609(2010).
[27]S. Y. Yang,Drug Discov. Today 15,444(2010).
[28]J. J. Irwin and B. K. Shoichet,J. Chem. Inf. Model. 45,177(2005).
[29]C. A. Lipinski,J. Pharmacol. Toxicol. Methods 44,235 (2000).
[30]P. S. Kutchukian,D. Lou,and E. I. Shakhnovich,J. Chem. Inf. Model. 49,1630(2009).
[31]P. S. Kutchukian and E. I. Shakhnovich,Exp. Opin. Drug Discovery 5,789(2010).
[32]R. A. Friesner,R. B. Murphy,and M. P. Repasky,L. L. Frye,J. R. Greenwood,T. A. Halgren,P. C. Sanschagrin,and D. T. Mainz,J. Med. Chem. 49,6177 (2006).
[33]M. B. Lazarus,Y. Nam,J. Jiang,P. Sliz,and S. Walker,Nature 469,564(2011).
[34]M. Fischer,R. G. Coleman,J. S. Fraser,and B. K. Shoichet,Nature Chem. 6,575(2014).
[35]H. C. Dorfmueller,V. S. Borodkin,D. E. Blair,S. Pathak,I. Navratilova,and D. M. van Aalten,Amino Acids 40,781(2011).
[36]JKlustor,http://www.chemaxon.com,ChemAxon:J. Chem. 11.2.0,(2015).
DOI:10.1063/1674-0068/29/cjcp1510211
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Impact of Eu3+Ions on Physical and Optical Properties of Li2O-Na2O-B2O3Glass
- Investigation of Ultrafast Electronic Transfer Process on Organic/Inorganic Heterojunction by Femtosecond Transient Absorption
- Ordered Toroid Structures of Nanoparticles in Self-attractive Semiflexible Polymer/Nanoparticle Composites
- Morphology and Growth Process of Bat-like ZnO Crystals by Thermal Evaporation
- Epitaxial Growth and Thermoelectric Measurement of Bi2Te3/Sb Superlattice Nanowires
- Controlled Synthesis of PCL/PVP Copolymer by RAFT Method and Its Hydrophilic Block-Dependent Micellar Behaviors