First-Principles Investigation on Triazine Based Thermally Activated Delayed Fluorescence Emitters
2016-07-05JianzhongFanShuaiQiuLiliLinChuankuiWangSchoolofPhysicsandElectronicShandongNormalUniversityJinan250014China
Jian-zhong Fan,Shuai Qiu,Li-li Lin,Chuan-kui WangSchool of Physics and Electronic,Shandong Normal University,Jinan 250014,China
First-Principles Investigation on Triazine Based Thermally Activated Delayed Fluorescence Emitters
Jian-zhong Fan,Shuai Qiu,Li-li Lin∗,Chuan-kui Wang∗
School of Physics and Electronic,Shandong Normal University,Jinan 250014,China
(Dated:Received on August 26,2015;Accepted on December 4,2015)
Three kinds of triazine based organic molecules designed for thermally activated delayed fluorescence(TADF)emitters are investigated by first-principles calculations. An optimal Hartree-Fork(HF)method is adopted for the calculation of energy gap between the first singlet state(S1)and the first triplet state(T1). The natural transition orbital,the electronhole(e-h)distribution and the e-h overlap diagram indicate that the S1 states for the three systems include both charge-transfer and some localized excitation component. Further quantitative analysis of the excitation property is performed by introducing the index∆r and the integral of e-h overlap S. It is found that symmetric geometry is a necessary condition for TADF emitters,which can provide more delocalized transition orbitals and consequently a small S1-T1 energy gap. Artful inserting aromatic groups between donors and acceptors can significantly enhance the oscillator strength. Finally,the energy state structures calculated with the optimal HF method is presented,which can provide basis for the study of the dynamics of excited states.
Key words:First-principles,Thermally activated delayed fluorescence,Charge-transfer states
∗Authors to whom correspondence should be addressed. E-mail:linll@sdnu.edu.cn,ckwang@sdnu.edu.cn
I. INTRODUCTION
Organic light emitting diodes(OLEDs)have attracted much attention recently since some metal-free thermally active delayed fluorescent(TADF)emitters with internal quantum efficiency attending to 100%are reported[1-3]. The TADF emitters break the rule that the internal quantum efficiency of organic molecules in OLED can't exceed 25%,and are regarded as the third generation organic electroluminescent molecular materials. One common character of these TADF emitters is that they are composed of electron-accepting groups (A)and electron-donating groups(D),which results in small energy gap between the singlet excited states(S)and the triplet excited states(T). Consequently,triplet excitons can up-convert to singlet excitons by the reverse intersystem crossing(RISC)process,thus the exciton utilization efficiency and the internal quantum efficiency are enhanced significantly. It is indicated that separating the spatial distribution of the highest occupied molecular orbital(HOMO)and the lowest unoccupied molecular orbital(LUMO)by introducing the donor groups and acceptor groups is an effective way to get a small S-T energy gap[4]. Nevertheless,the fluorescence rate is decreased due to the significant chargetransfer property of S1 in the D-A based systems. Thus a trade-off strategy is highly required in the design of TADF emitters. Now some researches found that the fluorescent rate can be enhanced by adding an aromatic bridge between D and A to increase the D-A separating length[5]. Furthermore,D-A-D type and A-D-A type based TADF emitters have also been designed in order to increase the fluorescent intensity[6,7].
In this work,the triazine based TADF emitters are studied. The optimal HF method will be used to calculate the S-T energy gap. Detail analysis of transition orbitals and the electron-hole(e-h)distribution as well as the e-h overlap will be carried out. By quantitatively analysis of the charge transfer distance and the integral of the e-h overlap for the S1 state,some design strategies for TADF emitters with high exciton utilization efficiency as well as high fluorescent rate will be provided. In the end,the energy state structure will be presented for further study of the dynamics of excited states.
II. COMPUTATIONAL DETAILS
In this work,the geometries of three molecules are optimized with the density functional theory(DFT)method at the B3LYP/6-31G∗level. The excitation energy for several low-lying excited states is calculated with the optimal HF methods[8]and the 6-31G∗basis set is used. All the calculations above are realized in theGaussian 09 program[9]. Further,the transition property such as the e-h distribution and the overlap of e-h of excited states are analyzed with the multifunctional wavefunction analyzer(Multiwfn)[10]. Here,we just list the main calculation steps. For more theoretical and computational details,one can refer to the manual of Multiwfn. In this work,the e-h distribution is studied to analyze the excitation property of molecules. The density distribution of holes ρholeand electrons ρelecan be gracefully defined as:
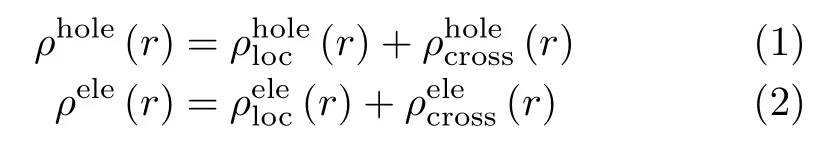
where“loc”and“cross”stand for the contribution of local term and cross term to the hole/electron distribution. They can be calculated with the formula as follows,

where φ denotes a molecular orbital(MO),and w is configuration coefficient. The label i and j run over all occupied MOs,l and m denote virtual MOs.
III. RESULTS AND DISCUSSION
In this work,three molecules composed of carbazol (D)and triazine(A)groups are theoretically studied.
2,4-bis(3-(9H-carbazol-9-yl)-9H-carbazol-9-yl)-6-phenyl-1,3,5-triazine[11-13],2-(12-phenylindolo(2,3-a)
carbazole-11-yl)-4-(3-(9H-carbazol-9-yl)-9H-carbazol-9-yl)-6-phenyl-1,3,5-triazine and 2,4-bis(3-(9H-carbazol-9-yl)-9-phenyl-9H-carbazole)-6-phenyl-1,3,5-triazine are marked with No.1,No.2,and No.3 respectively (shown in Fig.1). It is significant that No.1 is a typical D-A-D molecule,while No.2 is a D′-A-D type with different donor groups connected to the A group. No.3 is a generalized system of No.1,with an aromatic benzene ring inserted between D and A groups,which is a typical D-π-A-π-D molecule. The dihedral angles between D and A in three molecules optimized at B3LYP/6-31G∗level are different with each other (see Table I). In No.1,the angles between D and A are 19.79◦and 21.12◦. While the angles between D′and A in No.2 changed to 47.06◦and the D-A angle becomes 13.54◦. For No.3,the insert of the benzene ring between D and A makes the angles between the D group and the π unit become 51.40◦and 52.50◦. The significant increase of the angle between D and A will obstacle the charge transfer from D to A,thus less charge transfer will be expected for No.2 and No.3 in comparison with No.1. From Table I,we can also see that the insert of the benzene rings decreases the angle between the benzene ring and A(a5). In addition,the three benzene rings connected to the A group keep good planarity as illustrated in Fig.1.

TABLE I Dihedral angles(in(◦))of the ground states for three molecules optimized at the B3LYP/6-31G∗level. The labels involved are shown in Fig.1.

FIG. 1 Schematic diagram of geometric structures for three triazine based molecules. The dihedral angles of two surfaces are marked at the bonds connecting to them.
For TADF emitters,small energy gap between the S1 state and the T1 state is a necessary condition. Theoretical calculations of excited states usually adopt the time dependent density functional theory(TD-DFT)methods for large and medium systems. However,someresearches have shown that TD-DFT with nonhybrid functional always underestimates transition energies for charge-transfer(CT)states due to the neglecting of long-range Columbic attraction between the separated electrons and holes[14,15]. While TD-HF usually suffers from the so-called electron correlation problem and it may overestimate transition energies. It is found that the excited state calculation is largely depent on the HF%in functionals[16,17]. Therefore,an optimal HF%(OHF)in TD-DFT calculation should be determined. It has been proven that OHF is proportional to the CT amount q with a relationship of OHF=42q. The CT amount for No.1,No.2 and No.3 are 0.886,0.860 and 0.870 respectively(shown in Table II). Correspondingly,the OHF calculated for three molecules are 37.2,36.0 and 36.5. According to the results,the PBE38 (OHF=37.5)functional[18,19]is adopted to calculate the excitation energy for all the three molecules. For comparison,the calculation is also performed using the BMK(OHF=42)functional(see Table II). It is found that the excited energy is influenced significantly by the functional used. The excited energy calculated with the PBE38 functional for all the three molecules are smaller than that calculated with the BMK functional.
To calculate the zero-zero excited energy for the S1 and T1 states theoretically is quite time-consuming. Here,we adopt the useful formula as follows which has been proven correct and convenient[8].

here E0-0(S1)is the excitation energy for the S1 state. EVA(S1,OHF)is the vertical excitation energy of S1 calculated with an optimal HF functional.∆EVis the vibrational energy level difference between the 0-0 transition and the vertical transition.ω is 0.914 for our adopted functional(PBE38)tested by PhCz by the method proposed by the initial article[8]. To determine the value of∆EV,one should perform frequency calculation for S0 and S1 state respectively. Unfortunately the frequency calculation for excited state is quite timeconsuming and error-prone especially for these polyatomic molecules. Based on the research[8],the vertical S1 transition corresponds to the 0-1′transition for most CT compounds and∆EV=0.15 eV.∆Estrokes(Stokesshift energy loss)is assumed as 0.03 eV which excludes the contribution of solvation(~0.06 eV). In Eq.(8),EVA(T1,BLYP)is the vertical excitation energy for the T1 state calculated with the BLYP functional. With both Eq.(8)and Eq.(9),the excited energy of the first triplet state is calculated with the PBE38 functional as listed in Table III. It can be found that the value of E0-0(3CT)for No.1 is smaller than E0-0(3LE). However,for other two molecules,the value of E0-0(3LE)is relatively larger. It is indicated that the T1 states for No.1,No.2 and No.3 are CT,LE and LE state respectively. Thus the energy gaps between the S1 and T1 states for three molecules are predicted as shown in Table III. The energy gap of No.1 calculated with the PBE38 functional is 66.5 meV. For No.2 and No.3,the energy gaps are 194.4 and 448.4 meV respectively,both of which are much larger than that for No.1. The significant deviation of the energy gap is mainly induced by the geometry change and the difference of their excitation property.

TABLE II CT amount of S1 for three molecules. EVAin eV.
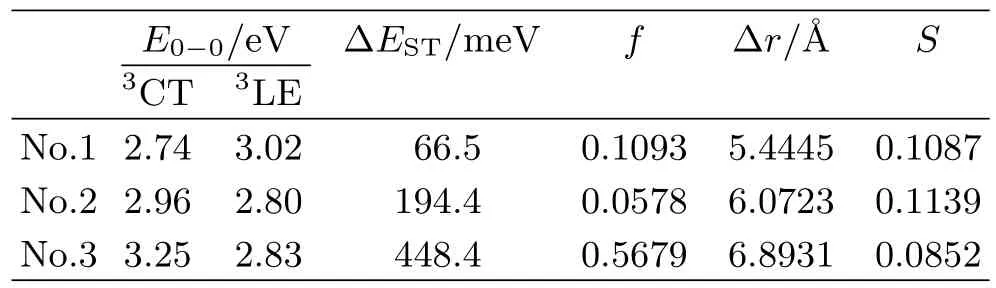
TABLE III Zero-zero excitation energy of the T1 state when the T1 state is a CT or LE state. Zero-zero energy gap between S1 and T1 calculated at the PBE38/6-31G∗level. The oscillator strength,the index∆r and S are also listed.
The natural transition orbitals(NTOs)and the e-h distributions of the S1 states for three molecules are shown in Fig.2(a)and(b). For No.1,holes are mainly located at two peripheral bicarbazole groups,while electrons are mainly distributed at the triazine and benzene groups. It means that electrons transfer from two D groups to the A group when the molecule is excited to the S1 state. It can also be confirmed by the NTO diagram,which shows that 96%of the excitation mainly happens from two D groups to the A group. Comparing No.2 with No.1,one can see that the replacement of one bicarbazole group with the indolocarbazole group breaks the symmetry of the molecule and also influences the excitation property. As illustrated,holes are only distributed on one bicarbazole group for No.2. Thus one can also deduce that electrons transfer mainly from one bicarbazole group to the triazine and benzene group in No.2,which is also in consistence with the NTO diagram. In comparison with the NTO of No.1,the transition orbital of No.2 is less delocalized. As mentioned above,the S-T energy gap of No.2 is much larger than that of No.1. This is consistent with the conclusion that the more delocalized the transition orbital is,the smaller the S-T energy gap will be[20]. For No.3,the transition happens from the two carbazole groups to the triazine group and the benzene groups between them. Consequently,significant charge transfer property for the S1 states of all the three molecules can be found. Nevertheless,there is still some component from localized excitation(LE)for the S1 state. In Fig.2(c),the overlap between electrons and holes can be directly seen. For No.1,the overlap mainly happens in the triazine group and two neighbor carbazole groups. It is similar for No.2,which is located at one carbazol group and the triazine group. For No.3,the benzene groups inserted between D and A groups are the main area for e-h overlap. As the oscillator strength of one molecule is proportional to the transition orbital overlap. The larger the e-h overlap is,the greater the transition orbital overlap will be,and the lager the oscillator strength will be. The fluorescent rate is also proportional to the oscillator strength. Thus a higher fluorescent intensity may be obtained for the system with larger e-h overlap. Nevertheless,large overlap of transition orbitals may also enlarge the S-T energy gap. Consequently,the trade-off of the CT and LE component in S1 will be important for highly efficient TADF emitters.
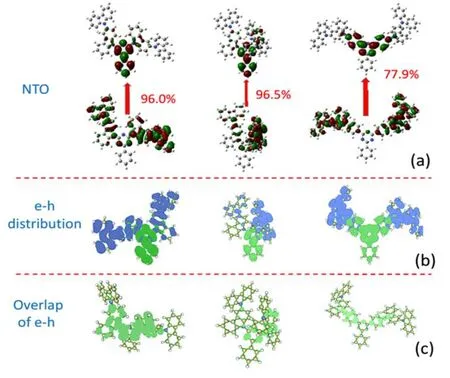
FIG. 2 (a)Natural transition orbitals(NTO)of S1 for three molecules. The percentage is the component of this kind of transition in all the transitions involved for S1.(b)and(c)present the electron-hole(e-h)distribution and the overlap of e-h for S1. The panel from left to right is for No.1,No.2,and No.3 respectively.
For quantitative comparison,the∆r index and the integral of the overlap of e-h(S)are introduced. The∆r index was proposed to measure charge-transfer length during electron excitation and the hole-particle pair interactions could be related to the distance covered during the excitations,and it is defined as follows[21]:
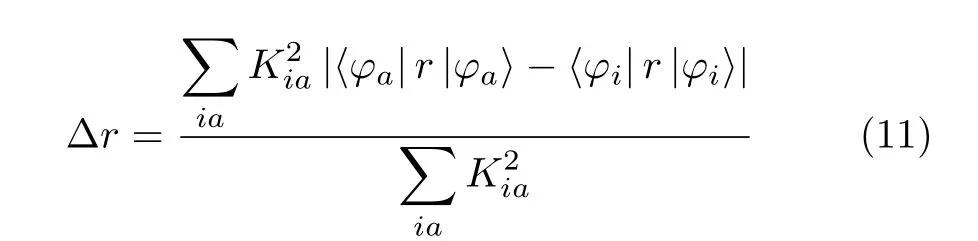
where the index a and i run over all the occupied MOs and virtual MOs respectively,Kiais the corresponding coefficients for the involved excitation. The smaller the∆r index is,the more likely the excitation is a LE mode. S is defined as the minimum of the electron density ρele(r)and hole density ρhole(r)everywhere. From Table III,one can see that the∆r index is larger than 2.0(the threshold value was proposed in the original paper of∆r)for all the three molecules,while the values of S are all very small. It further confirms that the S1 states for all the molecules are CT states. Besides,compared with No.1,No.3 possesses a smaller S (decreased by 22%)but a larger∆r(increased by 27%). That is to say,the insert of the aromatic groups between D and A groups may decrease the value of S to someextent but will increase the charge transfer distance. Both S and∆r has delicate relationship with the oscillator strength. In general,the larger the S value is,the stronger the oscillator strength will be. Similarly,the larger CT distance(∆r)can also induce greater oscillator strength. For the three molecules,the oscillator strengths(proportional to the fluorescent intensity)have been calculated with the PBE38 functional(see Table III). It is indicated that the oscillation strength of No.2 is smaller than that of No.1,while it becomes much larger for No.3. Despite that the introduction of the aromatic groups between D and A groups will enlarge the energy gap between S1 and T1,it can greatly enhance the oscillation strength,thus a larger fluorescent rate for No.3 is expected. According to the Einstein spontaneous emission rate formula,the radiation rate for No.3 calculated is 2.78×107s-1,which is a little larger than that of No.1(the fluorescent rate is 9.97×106s-1). This confirms that the insert of the aromatic groups between D and A groups maybe is a good way to enhance the fluorescent intensity. From the comparison of No.1 with No.2,we conclude that the energy gap between S1 and T1 can be minimized by adopting the symmetric geometry structure and the oscillator strength can also be enhanced by delocalizing the transition orbital. Consequently,a useful way to minimize the S-T energy gap and increase the fluorescent rate at the same time is to connect as many D groups as possible with the A group in the symmetric positions,and it has been proven efficient in many systems[22]. The calculation in this work suggest that appropriate arrangement of donor and acceptor moieties in triazine based molecules which is highly attractive for the construction of TADF emitters because of its electron deficient nature with three modification sites can allow both small S-T energy gap and feasible oscillator strength for highly efficiency TADF emitters.
The energy level structure and their dynamics determine the photophysical property of the molecules. The energy level structures of excited states for No.1,No.2 and No.3 are shown in Fig.3. All the calculations are performed with the optimal HF method at the PBE38/6-31G∗level. For No.1,the first triplet excited state(T1)and the second triplet excited state(T2)are degenerate. The energy of the third triplet excited state (T3)is about 72 meV higher than that of T2. All the three triplet states are lower in energy than the S1 state and the energy gap between S1 and T1 is 184 meV. As shown in the energy level diagram,S2 are much higher than S1. For No.2 and No.3,there are also three triplet states lower than S1 in energy. The gap between S1 and T1 is as large as 607 meV for No.2 and 856 meV for No.3. In comparison with the 0-0 energy gaps between S1(T1)and S0,the vertical excitation energy gaps are much larger. This also indicates that there is significant relaxation between S1(T1)and S0 in geometric structures. Generally,the internal conversion(IC)is much more quickly than the intersystem crossing(ISC) when the energy gap between the states with the same spin multiplicity is not large enough. We can deduce that the RISC process for all the three systems should mainly happens between the T1 state and the S1 state. Of course,the S2 state should also be in consideration when S2 is quasi-degenerate with S1 such as in No.2. However,the ISC process may mainly happen between the S1 state and the T3 state for both No.1 and No.2. For No.3,the ISC from S1 to T2 and T3 should be with the same importance. Based on the energy level structures,the theoretical model can be established and the dynamics of the excited states will be studied then. We will present our investigation on the dynamics of the excited states in the future work.

FIG. 3 Energy level structures of several lowest singlet excited states and triplet excited states for No.1,No.2,and No.3.
IV. CONCLUSION
In summary,first-principles investigations on three triazine based molecules designed for TADF emitters are performed. An optimal HF method with PBE38 functional is adopted to predict the S-T energy gap. The analysis of NTO,the e-h distribution and the eh overlap indicated that the S-T energy gap is closely related to the CT and LE component in S1. Quantitative calculation reflects that effective separation of electron and hole by inserting aromatic groups between D and A can significantly enhance the oscillator strength. Symmetric geometry is a necessary condition for TADF emitters,which can provide more delocalized transition orbitals and consequently a small S-T energy gap. The arrangement with D groups as many as possible connected with the A group may be a useful way to obtain both small S-T energy gap and large fluorescent rate,which is quite important for highly efficient TADF emitters. The geometry-property relationship will provide some enlightenment on the design of high efficient TADF emitters. The energy level structures calculated with the PBE38 functional will provide the basis for the study of the dynamics of the excited states.
V. ACKNOWLEDGMENTS
This work is supported by the National Natural Science Foundation of China(No.11374195 and No.21403133),Taishan Scholar Project of Shandong Province and the Scientific Research Foundation of Shandong Normal University,and the Promotive Research Fund for Excellent Young and Middle-aged Scientists of Shandong Province(No.BS2014CL001),and the General Financial Grant from the China Postdoctoral Science Foundation(No.2014M560571). Great thanks to Professor Yi Luo at USTC for his helpful suggestion and discussion in the detail calculation.
[1]M. A. Baldo,D. F. O'Brien,Y. You,A. Shoustikov,S. Sibley,M. E. Thompson,and S. R. Forrest,Nature 395,151(1998).
[2]J. H. Burroughes,D. D. C. Bradley,A. R. Brown,R. N. Marks,K. Mackay,R. H. Friend,P. L. Burns,and A. B. Holmes,Nature 347,539(1990).
[3]S. Reineke,F. Lindner,G. Schwartz,N. Seidler,K. Walzer,B. Lussem,and K. Leo,Nature 459,234 (2009).
[4]Y. Tao,K. Yuan,T. Chen,P. Xu,H. H. Li,R. F. Chen,C. Zheng,L. Zhang,and W. Huang,Adv. Mater. 26,7931(2014).
[5]Q. S. Zhang,H. Kuwabara,W. J. J. Potscavage,S. P. Huang,Y. Hatae,T. Shibata,and C. Adachi,J. Am. Chem. Soc. 136,18070(2014).
[6]M. Taneda,K. Shizu,H. Tanaka,and C. Adachi,Chem. Commun. 51,5028(2015).
[7]Q. S. Zhang,J. Li,K. Shizu,S. Huang,S. Hirata,H. Miyazaki,and C. Adachi,J. Am. Chem. Soc. 134,14706(2012).
[8]S. P. Huang,Q. S. Zhang,Y. Shiota,T. Nakagawa,K. Kuwabara,K. Yoshizawa,and C. Adachi,J. Chem. Theory Comput. 9,3872(2013).
[9]G. W. T. M. J. Frisch,H. B. Schlegel,G. E. Scuseria,M. A. Robb,J. R. Cheeseman,G. Scalmani,V. Barone,B. Mennucci,G. A. Petersson,H. Nakatsuji,M. Caricato,X. Li,H. P. Hratchian,A. F. Izmaylov,J. Bloino,G. Zheng,J. L. Sonnenberg,M. Hada,M. Ehara,K. Toyota,R. Fukuda,J. Hasegawa,M. Ishida,T. Nakajima,Y. Honda,O. Kitao,H. Nakai,T. Vreven,J. A. Montgomery,Jr.,J. E. Peralta,F. Ogliaro,M. Bearpark,J. J. Heyd,E. Brothers,K. N. Kudin,V. N. Staroverov,R. Kobayashi,J. Normand,K. Raghavachari,A. Rendell,J. C. Burant,S. S. Iyengar,J. Tomasi,M. Cossi,N. Rega,J. M. Millam,M. Klene,J. E. Knox,J. B. Cross,V. Bakken,C. Adamo,J. Jaramillo,R. Gomperts,R. E. Stratmann,O. Yazyev,A. J. Austin,R. Cammi,C. Pomelli,J. W. Ochterski,R. L. Martin,K. Morokuma,V. G. Zakrzewski,G. A. Voth,P. Salvador,J. J. Dannenberg,S. Dapprich,A. D. Daniels,O. Farkas,J. B. Foresman,J. V. Ortiz,J. Cioslowski,and D. J. Fox,Gaussian 09,Revision D.01,Wallingford CT,USA:Gaussian Inc.(2013).
[10]T. Lu and F. W. Chen,J. Comput. Chem. 33,580 (2012).
[11]T. Y. Sae Youn Lee,H. Nomura,and C. Adachi,Appl. Phys. Lett 101,093306(2012).
[12]Y. Z. Li,Y. Sun,Y. Q. Li,and F. C. Ma,Chin. J. Chem. Phys. 20,59(2007).
[13]Q. Li,F. Q. Huang,J. D. Hu,and K. Q. Zhao,Chin. J. Chem. Phys. 19,401(2006).
[14]A. Dreuw and M. Head Gordon,J. Am. Chem. Soc. 126,4007(2004).
[15]A. Dreuw and M. Head Gordon,Chem. Rev. 105,4009 (2005).
[16]P. Comba Ed.,Modeling of Molecular Properties,Weinheim:Wiley VCH,37(2011).
[17]C. A. Guido,P. Cortona,B. Mennucci,and C. Adamo,J. Chem. Theory Comput. 9,3118(2013).
[18]S. Grimme,J. Antony,S. Ehrlich,and H. Krieg,J. Chem. Phys. 132,154104(2010).
[19]L. Goerigk and S. Grimme,J. Chem. Phys. 132,184103 (2010).
[20]S. Hirata,Y. Sakai,K. Masui,H. Tanaka,S. Y. Lee,H. Nomura,N. Nakamura,M. Yasumatsu,H. Nakanotani,Q. Zhang,K. Shizu,H. Miyazaki,and C. Adachi,Nat. Mater. 14,330(2015).
[21]C. A. Guido,S. Knecht,J. Kongsted,and B. Mennucci. J. Chem. Theory Comput. 9,2209(2013).
[22]H. Uoyama,K. Goushi,K. Shizu,H. Nomura,and C. Adachi,Nature 492,234(2012).
DOI:10.1063/1674-0068/29/cjcp1508181
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Virtual Screening of Human O-GlcNAc Transferase Inhibitors
- Comparative Theoretical Studies on Several Energetic Substituted Dioxin-imidazole Derivatives
- Controlled Synthesis of PCL/PVP Copolymer by RAFT Method and Its Hydrophilic Block-Dependent Micellar Behaviors
- Epitaxial Growth and Thermoelectric Measurement of Bi2Te3/Sb Superlattice Nanowires
- Morphology and Growth Process of Bat-like ZnO Crystals by Thermal Evaporation
- Investigation of Ultrafast Electronic Transfer Process on Organic/Inorganic Heterojunction by Femtosecond Transient Absorption