Theoretical Study on Gas Phase Reactions of OH Hydrogen-Abstraction from Formyl Fluoride with Different Catalysts
2016-07-05DingmeiWngZhengwenLongXingfengTnBoLongWeijunZhngDeprtmentofPhysicsGuizhouUniversityGuiyng550025ChinCollegeofComputerndInformtionEngineeringGuizhouMinZuUniversityGuiyng550025ChinLortoryofEnvironmentSpectroscopyAnhuiIn
Ding-mei Wng,Zheng-wen Long,Xing-feng Tn,Bo Long∗,Wei-jun Zhng. Deprtment of Physics,Guizhou University,Guiyng 550025,Chin . College of Computer nd Informtion Engineering,Guizhou MinZu University,Guiyng 550025,Chin c. Lortory of Environment Spectroscopy,Anhui Institute of Optics nd Fine Mechnics,Chinese Acdemy of Sciences,Hefei 230031,Chin d. Key Lortory of Atmospheric Composition nd Opticl Rdition,Anhui Institute of Optics nd Fine Mechnics,Chinese Acdemy of Sciences,Hefei 230031,Chin
Theoretical Study on Gas Phase Reactions of OH Hydrogen-Abstraction from Formyl Fluoride with Different Catalysts
Ding-mei Wanga,Zheng-wen Longa,Xing-feng Tanb,Bo Longb∗,Wei-jun Zhangc, d∗
a. Department of Physics,Guizhou University,Guiyang 550025,China b. College of Computer and Information Engineering,Guizhou MinZu University,Guiyang 550025,China c. Laboratory of Environment Spectroscopy,Anhui Institute of Optics and Fine Mechanics,Chinese Academy of Sciences,Hefei 230031,China d. Key Laboratory of Atmospheric Composition and Optical Radiation,Anhui Institute of Optics and Fine Mechanics,Chinese Academy of Sciences,Hefei 230031,China
(Dated:Received on September 4,2015;Accepted on December 31,2015)
The mechanisms and kinetics of the gas phase reactions that the hydrogen atom in formyl fluoride(FCHO)abstracted by OH in the presence of water,formic acid(FA),or sulfuric acid(SA)are theoretically investigated at the CCSD(T)/6-311++G(3df,3pd)//M06-2X/6-311++G(3df,3pd)level of theory. The calculated results show that the barriers of the transition states involving catalysts are lowered to -2.89,-6.25,and -7.76 kcal/mol from 3.64 kcal/mol with respect to the separate reactants,respectively,which reflects that those catalysts play an important role in reducing the barrier of the hydrogen abstraction reaction of FCHO with OH. Additionally,using conventional transition state theory with Eckart tunneling correction,the kinetic data demonstrate that the entrance channel X···FCHO+OH (X=H2O,FA,or SA)is significantly more favorable than the pathway X···OH+FCHO. Moreover,the rate constants of the reactions of FCHO with OH radical with H2O,FA,or SA introduced are computed to be smaller than that of the naked OH+FCHO reaction because the concentration of the formed X···FCHO or X···OH complex is quite low in the atmosphere.
Key words:Formyl fluoride,Hydrogen abstraction,Reaction mechanisms,Rate constants
∗Authors to whom correspondence should be addressed. E-mail:wwwltcommon@sina.com,wjzhang@aiofm.ac.cn
I. INTRODUCTION
Formyl halides are important reactive molecules,which are formed via the atmospheric degradation intermediates of several halocarbon,for example,chlorofluorocarbons(CFCs)and hydrochlorofluorocarbons (HCFCs)[1-3]. Although the CFCs,HCFCs,and halogen are released into atmosphere from anthropogenic sources[4,5]at low concentrations,the proposal antarctic-like ozone hole shows that the implementation of the Montreal protocol has few positive effects on restricting CFCs and FCFCs to break the ozone and reduce the stratospheric halogen loading [5,6]. Therefore,it is of great necessity and importance to study the atmospheric effects and lifetimes of halogenated organic compounds. Formyl fluoride (FCHO)is one of the formyl halides in the upper troposphere,which is a major oxidation product of HFC-134a (CF3CFH2)and HFC-41(CH3F)[1-7]. For example,the OH radical reaction with HFC-134a in the presence of NOxleads to generate FCHO with a yield of 70%[7]. Thus,exploring the atmospheric oxidation processes of FCHO is required to fully estimate the effects of atmospheric environment of halogenated organic compounds.
Hydroxyl radical(OH)is one of the dominant reactive species in both polluted troposphere and nature,which plays an important role in the atmospheric chemistry[8]. There have been some experimental and theoretical investigations on the reaction of OH with FCHO[9-12],which indicates that the formation of FCO+H2O via hydrogen atom abstraction is the most feasible channel,whereas the formation of FCOOH+H via OH addition to the central carbon atom in the carbonyl oxide is negligible. Moreover,the rate constant of the reaction of FCHO and OH radical is calculated to be 4×10-15cm3/(molecule·s)[12],which shows the atmospheric lifetime of FCHO with respect to reaction with OH is a lower limit of 10 years. On the other hand,experimental and theoretical studies have revealed that the single water molecule could make a contribution to decreasing the reaction barriers in the homogeneous phase or heterogeneous processes[13-25]. In addition,atmospheric acids such as formic acid(FA)[26-28]and sulfuric acid(SA)[29-31]have been theoretically proven to reduce the barriers in the hydrolysis reaction [26,30-35]and isomerization processes[36-39]. The-refore,it is of great necessity to investigate the FCHO+OH reaction in the presence of H2O,FA,or SA.
In the present work,in order to explore the reaction mechanisms and kinetics for the hydrogen abstraction of the title reactions and determine whether these reactions are of great importance in the atmosphere,we consider that H2O,FA,and SA catalyzes gas-phase reaction of FCHO with OH using ab initio methods and conventional transition state theory(TST). The present findings not only evaluate the relatively catalytic abilities of the water,FA and SA based on the detailed potential energy surface,but also provide new insights into atmospheric oxidation processes of FCHO. The present results could have potential applications in understanding and elucidating the homogenous reaction processes of FCHO in the atmosphere.
II. COMPUTATIONAL METHODS
The calculations herein were performed using the Gaussian 09 program package[40]. Firstly,the geometrical structures of all the reactants,transition states,complexes,and products were optimized at the M06-2X/6-311++G(3df,3pd)level of theory. The M06-2X method is a highnonlocality functional with double the amount of nonlocalexchange(2X)[41],which has been shown to be sufficiently reliable for predicting geometries and frequencies of the stationary points [30,42-44]. At the same level,the corresponding frequency calculations of the optimized geometries were also done to prove the characters of minima without imaginary frequency and the transition state with only one imaginary frequency. We also performed the intrinsic reaction coordinate(IRC)[45]calculations at M06-2X/6-311++G(3df,3pd)level of theory in order to determine if a given transition state connects with the desired reactant and product.
Secondly,to refine relative energies,the single point energies were also calculated at the CCSD(T)/6-311++G(3df,3pd)level based on the M06-2X/6-311++G(3df,3pd)optimized geometries using Molpro software[46]. In these calculations,we also took into account the T1 diagnostic[47,48]in the CCSD wave function to determine the reliability of single-determinantbased methods. According to Rienstra-Kiracofe[48],the CCSD(T)calculation is considered to be reliable when the diagnostic values of the T1 of the closed-shell and open-shell species do not exceed the threshold of 0.02 and 0.044,respectively. Additionally,the basis set superposition error(BSSE)for the complexes was evaluated using the counterpoise method by Boys and Bernardi[49]at the M06-2X/6-311++G(3df,3pd)level of theory and the effect of spin-orbit coupling for the OH radical(-0.20 kcal/mol)[50]are considered in this method.
Finally,the rate constants of these elementary reactions were estimated using conventional TST[51-55]with Eckart[56]tunneling correction applied widely in atmospheric reactions in the literature[14,30,57-64],which were performed in the TheRate program[65]. As shown in Table I,the T1 diagnostic values of all species do not extend the upper limit in this work,which indicates that the CCSD(T)calculations are reliable.
III. RESULTS AND DISCUSSION
A. The reaction of FCHO with OH assisted by H2O
In order to demonstrate the reliability of theoretical methods utilized herein and better estimate the catalytic roles of H2O,FA,and SA,we also re-investigate the hydrogen abstraction and addition reaction of FCHO with OH. We find two reaction channels of are hydrogen abstraction and radical addition,which are similar to the OH+HCHO reaction[15,66-68]. As shown in Fig.1 and Table I,the barriers of the hydrogen abstraction channel is 5.09 kcal/mol relative to prereactive complex CR1,which is in good agreement with the value of 6.0 kcal/mol[9]. Therefore,the results show that the theoretical methods used herein are reliable for the OH+FCHO reaction. Additionally,due to the high energy barrier of OH addition to the carbon atom in FCHO,the addition reaction channel is negligible in the atmosphere.
The reactants are FCHO and the OH···H2O complex or OH and the FCHO···H2O complex in the reaction of OH with FCHO with a single water molecule being added,as shown in Fig.2. The capital letter“W”has been added to denote a single water molecule involved in the OH+FCHO reaction. The reaction between FCHO and OH catalyzed by H2O occurs through the pre-reactive complex CRW before the transition state TSW and the post-reactive complex CPW,leading to the formation of FCO+2H2O. The process is similar to the cases of HCHO+OH[15],HOCl+OH[14],and H2O2+OH[69]in the presence of water. From Table I,the binding energy of the H2O···OH is found to be -3.52 kcal/mol,which is in good agreement with the previous reported values of -3.77[70],-3.97[71],and -3.60 kcal/mol[72]. It is worth noting that CRW has a seven-membered-ring structure,where FCHO acts as both hydrogened-bond accepter and donor simultaneously. Additionally,the transition state TSW has an approximately planar configuration,where the breaking C1···H1 bond is elongated to 1.22Å from the equilibrium bond length of 1.09Å,and the bond length of H1···O2 is 1.01Å shorter than that of CRW. In Table I,the binding and activation energies of the CRW and TSW are -7.39 and -2.89 kcal/mol with BSSE correction relative to the reactants,respectively. The barrier of TSW is estimated to be 4.5 kcal/mol relative to the pre-reactive complex CRW,which is about 0.5 kcal/mol lower than that of the naked OH+FCHOreaction. Thus,H2O can play a positive role in the reduction of barrier of OH+FCHO. Moreover,the free energy calculated at the CCSD(T)/6-311++G(3df,3pd)//M06-2X/6-311++G(3df,3pd)level of theory is -18.66 kcal/mol,which indicates that the reaction process is thermodynamically favorable.

TABLE I The enthalpies(∆H),Gibbs free energies(∆G),and relative energies(∆E)of all species for the OH hydrogen-abstraction from FCHO with H2O,HCOOH,or H2SO4with zero-point energy correction included(FA=HCOOH,SA=H2SO4)a.
B. The reaction of FCHO with OH assisted by FA and SA
FA catalyzes the reaction of FCHO with OH via the possible entrance channels with FA···OH+FCHO or FA···FCHO+OH acting as reactants. Because formic acid has two different oxygen atoms,we find four reaction channels RF,RFa,RFb,and RFc for the reaction of the hydrogen atom of FCHO abstracted by OH and the corresponding transition states TSF,TSFa,TSFb,and TSFc,leading to the formation of the products FCO+H2O+HCOOH as shown in Fig.3 and Fig.S(a)-(c)(in supplementary materials). The pre-reactive complexes CRF,CRFa,CRFb,and CRFc are -10.17,-7.84,-7.14,and -5.06 kcal/mol in Table I and Table S1(in supplementary materials),respectively,which shows that the most stable one is the nine-membered-ring complex CRF. Therefore,we mainly discuss the reaction channel as presented in Fig.3. The reaction starts with two channels FA···OH+FCHO or FA···FCHO+OH,which leads to the formation of pre-reactive complex CRF,pro-ceeding to the transition state TSF before the formation of post-reactive complex CPF,and subsequently forming FCO+H2O+HCOOH. As shown in Table I,the binding energy of the HCOOH···OH is -3.41 kcal/mol at the CCSD(T)/6-311++G(3df,3pd)//M06-2X/6-311++G(3df,3pd)level of theory with the BSSE correction and considering the2P3/2spin-orbit state of OH radical,which is in good agreement with the previous values -3.68 kcal/mol[73]and -2.77 kcal/mol[15]. In CRF,both the carbonyl and the hydroxyl groups in FA are involved in the ring configuration,where there are three hydrogen bondings. In TSF,the bond lengths of C1···H1 and H1···O2 are 1.23 and 1.26Å,respectively,which are similar to those of the TSW. It is noted that the geometrical parameters of the two hydrogen bondings in TSF have little difference from those in CRF. The energy barrier is computed to be 3.92 kcal/mol with respect to CRF in Table I,which is about 1 kcal/mol lower than that of OH+FCHO. Moreover,the barrier is lower about 0.5 kcal/mol than that of OH+FCHO reaction in the presence of water. Thus,the results reflect that FA exerts a strong catalytic role in lowering the barrier of OH+FCHO. Additionally,the barriers of TSFa,TSFb,and TSFc in Table S1(in supplementary materials)are computed to be 4.41,5.27,and 4.25 kcal/mol relative to the respective pre-reactive complex,which is higher than that of TSF.
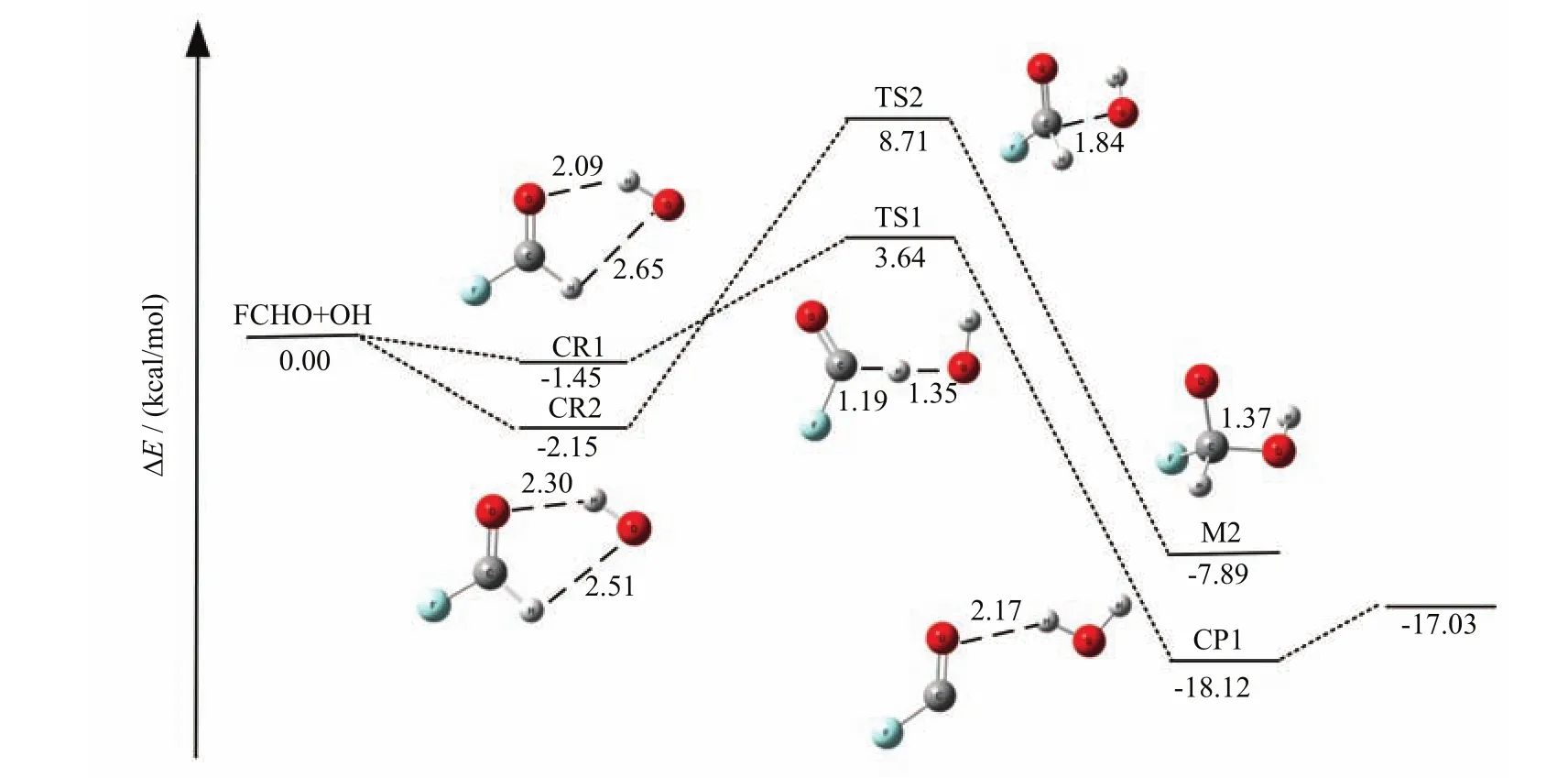
FIG. 1 Schematic potential energy surface for the hydrogen abstraction and addition reaction of FCHO with OH radical at the CCSD(T)/6-311++G(3df,3pd)//M06-2X/6-311++G(3df,3pd)level. The bond lengths are inÅ.
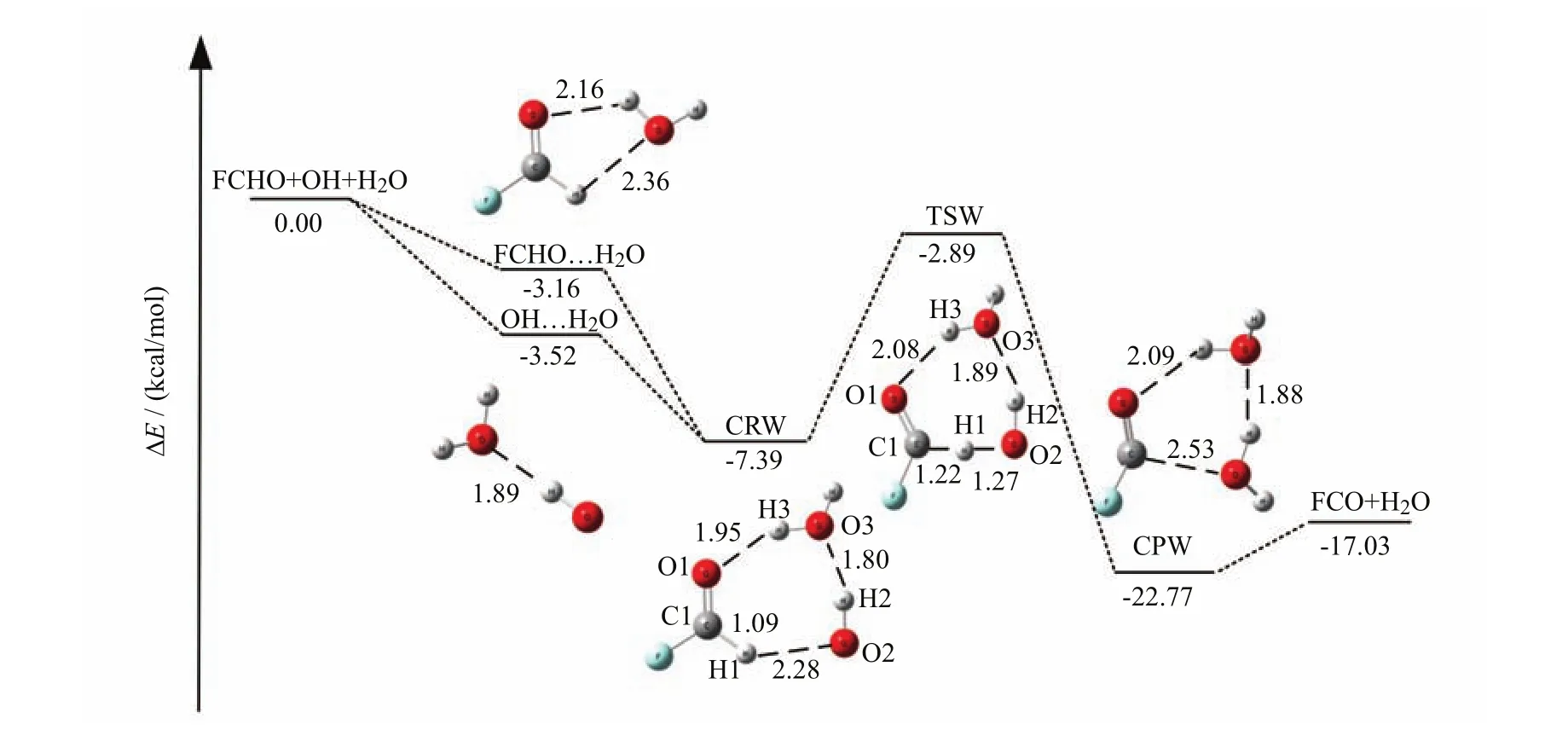
FIG. 2 Schematic potential energy surface for the hydrogen abstraction of FCHO with OH radical in the presence of H2O at the CCSD(T)/6-311++G(3df,3pd)//M06-2X/6-311++G(3df,3pd)level. The bond lengths are inÅ.
Regarding the reaction between FCHO and OH catalyzed by SA(Fig.4),the possible entrance pathways OH···H2SO4complex and FCHO···H2SO4complex can be formed. This reaction pathway undergoes the pre-reactive complex CRS before the transi-tion states TSS and the post-reactive complex CPS to form the corresponding products FCO+H2O+H2SO4. From Table I,the binding energy of the CRS is -11.02 kcal/mol with BSSE correction relative to the reactants at the CCSD(T)/6-311++G(3df,3pd)//M06-2X/6-311++G(3df,3pd)level of theory,which is 5.03 kcal/mol higher than that of the reaction of HCHO and OH assisted SA at the CCSD(T)/6-311++G(3df,3pd)//MP2(full)levels of theory[15]. Likewise,TSS possesses twisted nine-membered-ring configuration with the activation energy -7.76 kcal/mol relative to the free reactants.
From the discussion mentioned above,it can be seen to see that the barriers for the hydrogen abstraction process of FCHO assisted by water,FA,or SA are 4.50,3.92,and 3.26 kcal/mol respect to the pre-reactive complexes,respectively,which are semblable to the value of 5.09 kcal/mol in the reaction of FCHO with OH without catalyst. However,our calculations reveal that the activation energies are reduced to -2.89,-6.25,and -7.76 kcal/mol relative to free reactants in the presence of water,FA,and SA,respectively,the energy barriers of the reactions with the catalysts can be decreased,in comparison with the naked OH+FCHO reaction. Thus,a further kinetic study should be done to evaluate the possible catalytic effects of H2O,FA,and SA in the atmosphere.

FIG. 3 Schematic potential energy surface for the hydrogen abstraction of FCHO with OH radical catalyzed by HCOOH at the CCSD(T)/6-311++G(3df,3pd)//M06-2X/6-311++G(3df,3pd)level. The bond lengths are inÅ.

FIG. 4 Schematic potential energy surface for the hydrogen abstraction of FCHO with OH radical catalyzed by H2SO4at the CCSD(T)/6-311++G(3df,3pd)//M06-2X/6-311++G(3df,3pd)level. The bond lengths are inÅ.
C. Kinetics and potential applications in atmospheric chemistry
Based on the analysis of energies above,the function of water,FA,or SA is to lower the barrier for the hydrogen abstraction of the FCHO with OH reaction,which plays an important role in the formationof FCO+H2O. In order to examine the possible atmospheric impacts of these reactions discussed herein,we have also computed the rate constants of the reactions using conventional TST on the basis of the CCSD(T)/6-311++G(3df,3pd)//M06-2X/6-311++G(3df,3pd)potential energy surfaces discussed above over the temperature of 200-300 K. The reaction mechanisms of the reactions start with the formation of the pre-reactive complexes before the transition states and release the products.
For the naked reaction of FCHO with OH,the reaction mechanism can be characterized by the following reaction:

where k1and k-1express the forward and reverse rate constants for the first step,which forms the pre-reactive complex,k2corresponds to the products come into being for the second step. On the basis of the steady-state conditions,the overall rate constant is expressed as:

If k2is considerably smaller than k-1,the rate constant can be further simplified as:

where keqrepresents the equilibrium constant of the first step. The keqand k2can be calculated via the following:
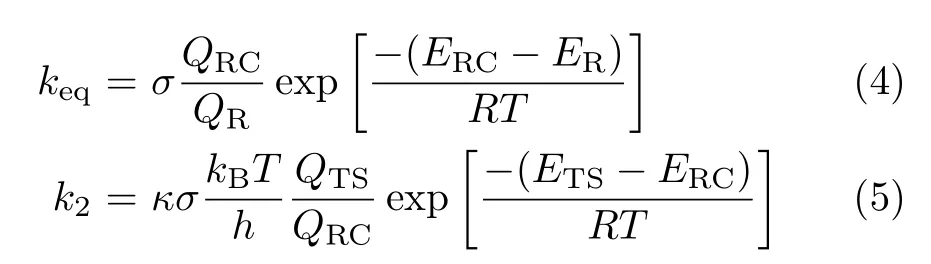
where QR,QRC,and QTSstand for the partition functions of the reactants,the pre-reactive complex,and the transition state,respectively.σ is the symmetry,κ is the transmission coefficient,kBis the Boltzmann constants,h is the Planck constants and R is the ideal gas constants. The ER,ERC,and ETSdenote the total energies of the reactants,pre-reactive complex,and transition state with ZPE correction.
As shown in Table II,it can be seen that the rate constant of the naked reaction FCHO+OH is in the range of 4.37×10-15cm3/(molecule·s)to 1.95×10-14cm3/(molecule·s)in the temperature of 200-300 K,which is in good agreement with the previous report by Timothy et al.[12](about 4×10-15cm3/(molecule·s)). From the calculated data,we can obtain a conclusion that the rate constants increase with temperature.

TABLE II The computational equilibrium constants(keqin molecules/cm3)and rate constants(cm3/(molecule·s))of reaction of the FCHO+OH without catalyst in temperature range of 200-300 K.
For the reactions of FCHO with OH catalyzed by water,FA or SA,which can proceed via two entrance channels:one is X···OH+FCHO and the other is X···FCHO+OH. The main steps of the two pathways can be considered as(where X=H2O,HCOOH,or H2SO4):

The reaction sequences are representatively regarded as one involving the formation of the CRX pre-reactive collision complex by means of Sinha[74]and Fliegl[75],which then lead to the unimolecular reaction.
In a general way,Eq.(8)is viewed as the most crucial step to determine the rate constant,and the rate can be shown as:

Assuming CRX is in equilibrium with the reactants and according to the steady-state conditions,similar to the gas phase reaction FA-catalyzed hydrolysis of SO3[74],the rate constant of the X···OH+FCHO pathway can be represented as:
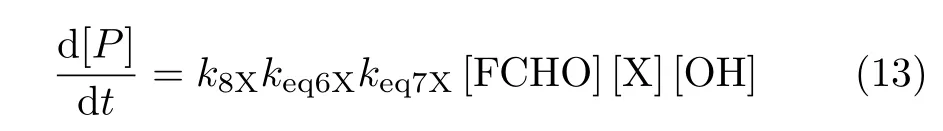
Consequently,the overall reaction rate constant is carried out by the following expression:

where keq6Xand keq7Xrepresent the equilibrium constants for Eq.(6)and Eq.(7),respectively,k8Xis the rate constant of Eq.(8),and[X]is the troposphere concentration of water,FA,or SA.
TABLE III The rate constantsand(cm3/(molecule·s))for the individual reaction pathway of the two channels for catalytic reaction with the temperature range 200-300 K(where X=H2O,HCOOH,or H2SO4).
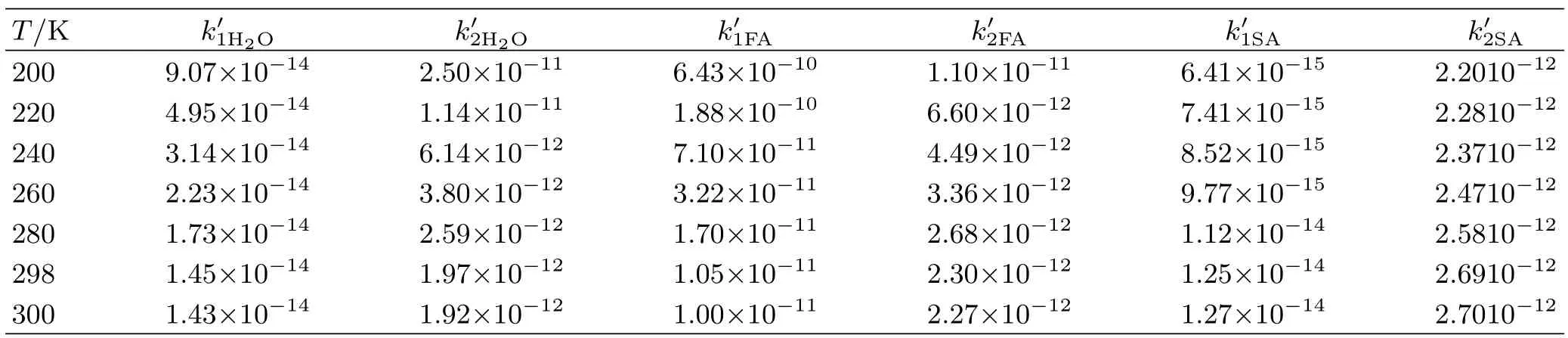
TABLE III The rate constantsand(cm3/(molecule·s))for the individual reaction pathway of the two channels for catalytic reaction with the temperature range 200-300 K(where X=H2O,HCOOH,or H2SO4).
T/K k′1H2O k′2H2O k′1FA k′2FA k′1SA k′2SA 200 9.07×10−14 2.50×10−11 6.43×10−10 1.10×10−11 6.41×10−15 2.2010−12220 4.95×10−14 1.14×10−11 1.88×10−10 6.60×10−12 7.41×10−15 2.2810−12240 3.14×10−14 6.14×10−12 7.10×10−11 4.49×10−12 8.52×10−15 2.3710−12260 2.23×10−14 3.80×10−12 3.22×10−11 3.36×10−12 9.77×10−15 2.4710−12280 1.73×10−14 2.59×10−12 1.70×10−11 2.68×10−12 1.12×10−14 2.5810−12298 1.45×10−14 1.97×10−12 1.05×10−11 2.30×10−12 1.25×10−14 2.6910−12300 1.43×10−14 1.92×10−12 1.00×10−11 2.27×10−12 1.27×10−14 2.7010−12

TABLE IV The ratio of rate constants of the two channels for the reactions of FCHO with OH radical catalyzed by water,FA,or SA with the temperature range 200-300 K.
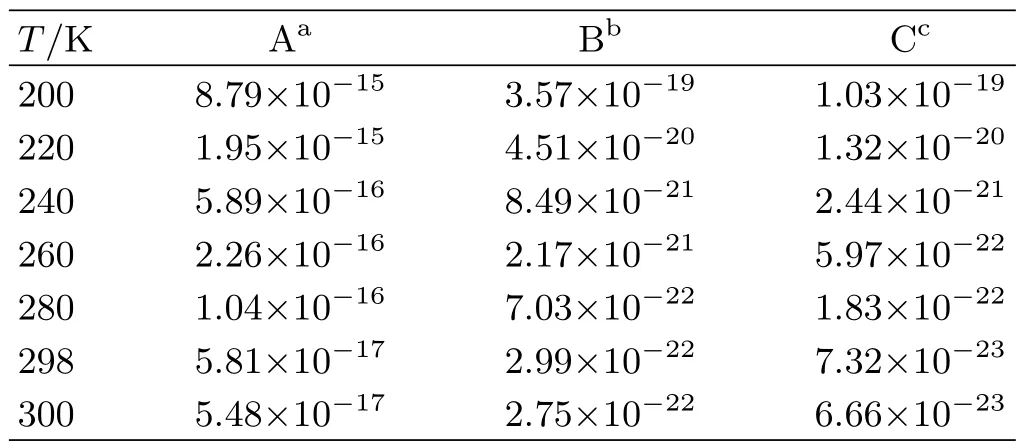
TABLE V Calculated rate constants(cm3/(molecule·s))of the more favorable channels for the reaction of FCHO+OH with water,FA,or SA.
Analogously,the rate constant of the pathway X···FCHO+OH can be written as(keq9Xand keq10Xdenote the equilibrium constants for Eq.(9)and Eq.(10):

If we do not think about Eq.(6)and(9),the rate constants for the title reaction assisted by catalyst can be expressed as:
In order to compare which entrance channel is more feasible,we calculated the ratio of the rate of the two reaction channels by the following equation:

For the H2O-assisted the hydrogen abstraction of FCHO by OH,the equilibrium constant,the rate constant,and the ratio of the two entrance channels in the temperature of 200-300 K are presented in Table S2(in supplementary materials). Additionally,the concentration of water molecule at 100%relative humidity is 7.70×1017molecules/cm3[76,77]. From Table III,the computational data indicates that the rate constantandare larger than that of the OH+FCHO reaction. Especially,the X(X=H2O and SA)···FCHO+OH reaction is faster than the reaction of FCHO with OH by 2-4 orders of magnitude when the formation process of the H2O···FCHO complex is ignored,but theis essentially the same as theHowever,as is shown in Table S2(in supplementary materials),when the formation process of the H2O···OH and H2O···FCHO complex in Eq.(6)and Eq.(9)is considered,the rate constants kH2O···OH+FCHO and kH2O···FCHO+OH are 3 orders of magnitude smaller than that of the naked OH+FCHO reaction at 298 K. It is noted that all the rate constants decrease with the increase in temperature. From Table IV,our calculations show that the reaction starting from H2O···FCHO complex with OH radical is more feasible. Considering the reactions assisted by formic acid and sulfur acid,the corresponding kinetic data are given in Table S3 and Table S4 (in supplementary materials),respectively. From the computational results presented herein,we obtain similar conclusions that the rate constants for Eq.(6)and Eq.(9)ingored are larger and the rate constants involv-ing those reaction steps of Eq.(6)and Eq.(9)are smaller than those of the reaction without catalyst. Specially,the Table IV also tells us that the rate value of the two pathways for formic acid assisted are almost the same,whereas the kH2SO4···FCHO+OHare about 3 orders of magnitude larger than kH2SO4···OH+FCHOin the temperature of 200-300 K. Therefore,the channel involving the H2SO4···FCHO with OH radical is faster than the other.
Based on the analysis above,the rate constants of the more favorable entrance channels for the FCHO+OH reaction assisted by water,FA,or SA are listed in Table V. The atmospheric concentrations of water,FA and SA are 7.70×1017,2.00×1011,and 4.00×108molecules/cm3,respectively[78,79]. Considering the high atmospheric concentrations of water,this simple relative rate analysis demonstrate that the hydrogen abstraction reaction of FCHO with OH catalyzed by water is more favorable and much faster than the FA-assisted or SA-assisted. As shown in Table V,the rate constant calculated at 298 K for the reaction assisted by water is 5.81×10-17cm3/(molecule·s),which is smaller by about 3 orders of magnitude than the reaction without catalyst. From the point of the view of dynamics,the hydrogen abstraction process of FCHO with OH radical catalyzed by water is of minor importance in the gas phase atmospheric chemistry.
IV. CONCLUSION
The mechanisms and kinetics of the gas-phase hydrogen abstraction reaction of FCHO with OH radical catalyzed by water,FA,or SA are theoretically investigated at the CCSD(T)/6-311++G(3df,3pd)//M06-2X/6-311++G(3df,3pd)level of theory. From the analysis of the calculational results,the participation of the a single water,FA,or SA plays an important role in the formation of FCO and H2O,which dramatically decrease the energy barriers of the corresponding transition state about 6.53,9.89,and 11.40 kcal/mol relative to that of the without catalyst,respectively. Additionally,based on the kinetic calculations,the reaction channel starting with X···FCHO+OH reactant is more favorable than the pathway involving X···OH+FCHO reactant. According to the calculations of the theoretical rate constants for these major reaction pathways with catalyst,we find that water has stronger catalysis than others. Moreover,the rate constants for these major reaction pathways with catalyst are much smaller than that of the reaction without any catalyst,which means that contribution of the catalysts water,FA,or SA is be of no account for the hydrogen abstraction reaction of FCHO+OH in the atmosphere. Even so,our study might be of great use to understand the atmospheric chemistry and estimate the other atmospheric oxidation processes which were assisted by water,FA,or SA.
Supplementary materials:Figure S(a),S(b),and S(c)show the potential energy profile for other three possible reaction channels of the reaction of FCHO with OH radical catalyzed by SA. Table S1 presents the enthalpies,Gibbs free energies,and relative energies of all species for other three possible reaction channels of the reaction FCHO+OH+HCOOH(RFa,RFb,and RFc)with zero-point energy correction included (in kcal/mol). Table S2,Table S3,and Table S4 show the equilibrium constants(molecules/cm3),the rate constants(cm3/molecule·s)for the individual reaction pathway and the ratio of rate expressions of the two channels for water,FA,or SA catalytic reactions with the temperature range of 200-300 K,respectively.
V. ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China(No.41165007 and No.21163003),the Science and Technology Foundation of Guizhou Province of China(No.[2011]2107 and No.[2012]2189),and the Science and Technology Foundation of Guizhou Province of Guizhou Minzu University of China(No.[2014]7380).
[1]A. S. Hasson,C. M. Moore,and I. W. M. Smith,Int. J. Chem. Kinet. 30,541(1998).
[2]E. Sanhueza and J. Heicklen,J. Phys. Chem. 79,7 (1975).
[3]J. Sehested and T. J. Wallington,Environ. Sci. Technol. 27,146(1993).
[4]G. L. Manney,L. Froidevaux,J. W. Waters,R. W. Zurek,W. G. Read,L. S. Elson,J. B. Kumer,J. L. Mergenthaler,A. E. Roche,A. O'Neill,R. S. Harwood,I. Mackenzie,and R. Swinbank,Nature 370,429(1994).
[5]P. A. Newman,L. D. Oman,A. R. Douglass,E. L. Fleming,S. M. Frith,M. M. Hurwitz,S. R. Kawa,C. H. Jackman,N. A. Krotkov,E. R. Nash,J. E. Nielsen,S. Pawson,R. S. Stolarski,and G. J. M. Velders,Atmos. Chem. Phys. 9,2113(2009).
[6]G. L. Manney,M. L. Santee,M. Rex,N. J. Livesey,M. C. Pitts,P. Veefkind,E. R. Nash,I. Wohltmann,R. Lehmann,L. Froidevaux,L. R. Poole,M. R. Schoeberl,D. P. Haffner,J. Davies,V. Dorokhov,H. Gernandt,B. Johnson,R. Kivi,E. Kyro,N. Larsen,P. F. Levelt,A. Makshtas,C. T. McElroy,H. Nakajima,M. C. Parrondo,D. W. Tarasick,P. von der Gathen,K. A. Walker,and N. S. Zinoviev,Nature 478,469(2011).
[7]T. J. Wallington,M. D. Hurley,J. C. Ball,and E. W. Kaiser,Environ. Sci. Technol. 26,1318(1992).
[8]J. Hoign´e and H. Bader,Water Res. 10,377(1976).
[9]N. Mora-Diez,J. R. Alvarez-Idaboy,and R. J. Boyd,J. Phys. Chem. A 105,9034(2001).
[10]B. S. Jursic,J. Mol. Struct:THEOCHEM 434,53 (1998).
[11]J. S. Francisco,J. Chem. Phys. 96,7597(1992).
[12]T. J. Wallington and M. D. Hurley,Environ. Sci. Technol. 27,1448(1993).
[13]B. Long,W. J. Zhang,X. F. Tan,Z. W. Long,Y. B. Wang,and D. S. Ren,J. Phys. Chem. A 115,1350 (2011).
[14]J. Gonzalez,J. M. Anglada,R. J. Buszek,and J. S. Francisco,J. Am. Chem. Soc. 133,3345(2011).
[15]W. Zhang,B. Du,and Z. Qin,J. Phys. Chem. A 118,4797(2014).
[16]B. Long,X. F. Tan,D. S. Ren,and W. J. Zhang,J. Mol. Struc:THEOCHEM 956,44(2010).
[17]A. J. C. Varandas,Int. J. Quantum. Chem. 114,1327 (2014).
[18]B. Long,W. J. Zhang,and Z. W. Long,Chin. J. Chem. Phys. 24,419(2011).
[19]T. Zhang,R. Wang,H. Chen,S. Min,Z. Wang,C. Zhao,Q. Xu,L. Jin,W. Wang,and Z. Wang,Phys. Chem. Chem. Phys. 17,15046(2015).
[20]B. Du and W. Zhang,Comput. Theor. Chem. 1049,90 (2014).
[21]X. Xu,R. P. Muller,and W. A. Goddard,Proc. Nati. Acad. Sci. 99,3376(2002).
[22]C. R. Chang,Y. G. Wang,and J. Li,Nano. Res. 4,131 (2011).
[23]Y. F. Zhao,Y. Yang,C. Mims,C. H. F. Peden,J. Li,and D. Mei,J. Catal. 281,199(2011).
[24]C. R. Chang,X. F. Yang,B. Long,and J. Li,ACS. Catal. 3,1693(2013).
[25]C. R. Chang,B. Long,X. F. Yang,and J. Li,J. Phys. Chem. C 119,16072(2015).
[26]B. Long,Z. W. Long,Y. B. Wang,X. F. Tan,Y. H. Han,C. Y. Long,S. J. Qin,and W. J. Zhang,ChemPhysChem 13,323(2012).
[27]S. Ghoshal and M. K. Hazra,RSC. Adv. 5,17623 (2015).
[28]M. Kumar,D. H. Busch,B. Subramaniam,and W. H. Thompson,J. Phys. Chem. A 118,9701(2014).
[29]J. Elm,M. Bilde,and K. V. Mikkelsen,J. Phys. Chem. A 117,6695(2013).
[30]B. Long,X. F. Tan,C. R. Chang,W. X. Zhao,Z. W. Long,D. S. Ren,and W. J. Zhang,J. Phys. Chem. A 117,5106(2013).
[31]M. Torrent-Sucarrat,J. S. Francisco,and J. M. Anglada,J. Am. Chem. Soc. 134,20632(2012).
[32]M. K. Hazra,J. S. Francisco,and A. Sinha,J. Phys. Chem. A 117,11704(2013).
[33]H. A. Rypkema,A. Sinha,and J. S. Francisco,J. Phys. Chem. A 119,4581(2015).
[34]M. K. Hazra,J. S. Francisco,and A. Sinha,J. Phys. Chem. A 118,4095(2014).
[35]M. K. Louie,J. S. Francisco,M. Verdicchio,S. J. Klippenstein,and A. Sinha,J. Phys. Chem. A 119,4347 (2015).
[36]R. J. Buszek,A. Sinha,and J. S. Francisco,J. Am. Chem. Soc. 133,2013(2011).
[37]S. Ghoshal and M. K. Hazra,J. Phys. Chem. A 118,4620(2014).
[38]A. Karton,Chem. Phys. Lett. 592,330(2014).
[39]L. Vereecken,D. R. Glowacki,and M. J. Pilling,Chem. Rev. 115,4063(2015).
[40]M. J. Frisch,G. W. Trucks,H. B. Schlegel,G. E. Scuseria,M. A. Robb,J. R. Cheeseman,G. Scalmani,V. Barone,B. Mennucci,G. A. Petersson,H. Nakatsuji,M. Caricato,X. Li,H. P. Hratchian,A. F. Izmaylov,J. Bloino,G. Zheng,J. L. Sonnenberg,M. Hada,M. Ehara,K. Toyota,R. Fukuda,J. Hasegawa,M. Ishida,T. Nakajima,Y. Honda,O. Kitao,H. Nakai,T. Vreven,J. A. Montgomery Jr.,J. E. Peralta,F. Ogliaro,M. Bearpark,J. J. Heyd,E. Brothers,K. N. Kudin,V. N. Staroverov,R. Kobayashi,J. Normand,K. Raghavachari,A. Rendell,J. C. Burant,S. S. Iyengar,J. Tomasi,M. Cossi,N. Rega,J. M. Millam,M. Klene,J. E. Knox,J. B. Cross,V. Bakken,C. Adamo,J. Jaramillo,R. Gomperts,R. E. Stratmann,O. Yazyev,A. J. Austin,R. Cammi,C. Pomelli,J. W. Ochterski,R. L. Martin,K. Morokuma,V. G. Zakrzewski,G. A. Voth,P. Salvador,J. J. Dannenberg,S. Dapprich,A. D. Daniels,¨O. Farkas,J. B. Foresman,J. V. Ortiz,J. Cioslowski,and D. J. Fox,Gaussian 09,Revision A. 02,Wallingford,CT:Gaussian,Inc.(2009).
[41]Y. Zhao and D. Truhlar,Theor. Chem. Account 120,215(2008).
[42]J. Elm,M. Bilde,and K. V. Mikkelsen,J. Chem. Theory. Comput. 8,2071(2012).
[43]M. Rooman and R. Wintjens,J. Biomol. Struct. Dyn. 32,532(2013).
[44]L. K. Sviatenko,L. Gorb,F. C. Hill,D. Leszczynska,S. I. Okovytyy,and J. Leszczynski,Chemosphere 134,31(2015).
[45]C. Gonzalez and H. B. Schlegel,J. Phys. Chem. 94,5523(1990).
[46]H. J. Werner,P. J. Knowles,G. Knizia,F. R. Manby,M. Sch¨utz,P. Celani,W. Gy¨orffy,D. Kats,T. Korona,R. Lindh,A. Mitrushenkov,G. Rauhut,K. R. Shamasundar,T. B. Adler,R. D. Amos,A. Bernhardsson,A. Berning,D. L. Cooper,M. J. O. Deegan,A. J. Dobbyn,F. Eckert,E. Goll,C. Hampel,A. Hesselmann,G. Hetzer,T. Hrenar,G. Jansen,C. K¨oppl,Y. Liu,A. W. Lloyd,R. A. Mata,A. J. May,S. J. Mc-Nicholas,W. Meyer,M. E. Mura,A. Nicklaß,D. P. O'Neill,P. Palmieri,D. Peng,K. Pfl¨uger,R. Pitzer,M. Reiher,T. Shiozaki,H. Stoll,A. J. Stone,R. Tarroni,T. Thorsteinsson,and M. Wang,MOLPRO,a package of ab initio programs(2012).
[47]T. J. Lee and P. R. Taylor,Int. J. Quantum Chem. Symp. 23,199(1989).
[48]J. C. Rienstra-Kiracofe,W. D. Allen,and H. F. Schaefer,J. Phys. Chem. A 104,9823(2000).
[49]S. F. Boys and F. Bernardi,Mol. Phys. 19,553(1970).
[50]J. Zheng,R. Meana-Pa˜neda,and D. G. Truhlar,J. Am. Chem. Soc. 136,5150(2014).
[51]D. G. Truhlar,B. C. Garrett,and S. J. Klippenstein,J. Phys. Chem. 100,12771(1996).
[52]M. G. Evans and M. Polanyi,Trans. Faraday. Soc. 31,875(1935).
[53]H. Eyring,J. Chem. Phys. 3,107(1935).
[54]W. M. Wei,R. H. Zheng,Y. K. Wu,F. Yang,and S. Hong,Chin. J. Chem. Phys. 27,659(2014).
[55]P. Wang,M. X. Yang,S. L. Zhang,S. P. Huang,and H. P. Tian,Chin. J. Chem. Phys. 26,35(2013).
[56]C. Eckart,Phys. Rev. 35,1303(1930).
[57]B. Long,X. F. Tan,Z. W. Long,Y. B. Wang,D. S. Ren,and W. J. Zhang,J. Phys. Chem. A 115,6559 (2011).
[58]B. Long,W. J. Zhang,X. F. Tan,Z. W. Long,Y. B. Wang,and D. S. Ren,Comput. Theor. Chem. 964,248(2011).
[59]J. R. Alvarez-Idaboy,N. Mora-Diez,and A. Vivier-Bunge,J. Am. Chem. Soc. 122,3715(2000).
[60]B. Long,C. R. Chang,Z. W. Long,Y. B. Wang,X. F. Tan,and W. J. Zhang,Chem. Phys. Lett. 581,26 (2013).
[61]F. Y. Liu,Z. W. Long,X. F. Tan,and B. Long,Comput. Theor. Chem. 1038,33(2014).
[62]F. Y. Liu,Z. W. Long,X. F. Tan,and B. Long,J. Mol. Model. 20,1(2014).
[63]M. Hajmalek,H. Aghaie,K. Zare,and M. Aghaie,Chin. J. Chem. Phys. 27,672(2014).
[64]C. F. Song,Z. M. Tian,Q. X. Lib,and T. J. He,Chin. J. Chem. Phys. 22,87(2009).
[65]W. T. Duncan,R. L. Bell,and T. N. Truong,J. Comput. Chem. 19,1039(1998).
[66]B. D'Anna,V. Bakken,J. Are Beukes,C. J. Nielsen,K. Brudnik,and J. T. Jodkowski,Phys. Chem. Chem. Phys. 5,1790(2003).
[67]J. R. Alvarez-Idaboy,N. Mora-Diez,R. J. Boyd,and A. Vivier-Bunge,J. Am. Chem. Soc. 123,2018(2001).
[68]Y. Zhao,B. Wang,H. Li,and L. Wang,J. Mol. Struc-THEOCHEM 818,155(2007).
[69]R. J. Buszek,M. Torrent-Sucarrat,J. M. Anglada,and J. S. Francisco,J. Phys. Chem. A 116,5821(2012).
[70]J. Gonzalez and J. M. Anglada,J. Phys. Chem. A 114, 9151(2010).
[71]M. A. Allodi,M. E. Dunn,J. Livada,K. N. Kirschner,and G. C. Shields,J. Phys. Chem. A 110,13283(2006).
[72]P. Soloveichik,B. A. O'Donnell,M. I. Lester,J. S. Francisco,and A. B. McCoy,J. Phys. Chem. A 114,1529 (2010).
[73]A. Galano,J. R. Alvarez-Idaboy,M. E. Ruiz-Santoyo,and A. Vivier-Bunge,J. Phys. Chem. A 106,9520 (2002).
[74]M. K. Hazra and A. Sinha,J. Am. Chem. Soc. 133,17444(2011).
[75]H. Fliegl,A. Gl¨oß,O. Welz,M. Olzmann,and W. Klopper,J. Chem. Phys. 125,054312(2006).
[76]J. Marti and K. Mauersberger,Geophys. Res. Lett. 20,363(1993).
[77]W. Wagner and A. Pruß,J. Phys. Chem. Ref. Data. 31,387(2002).
[78]P. L. Hanst,N. W. Wong,and J. Bragin,Atmos. Environ. 16,969(1982).
[79]S. Mikkonen,S. Romakkaniemi,J. N. Smith,H. Korhonen,T. Pet¨aj¨a,C. Plass-Duelmer,M. Boy,P. H. Mc-Murry,K. E. J. Lehtinen,J. Joutsensaari,A. Hamed,R. L. Mauldin Iii,W. Birmili,G. Spindler,F. Arnold,M. Kulmala,and A. Laaksonen,Atmos. Chem. Phys. 11,11319(2011).
DOI:10.1063/1674-0068/29/cjcp1509187
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Virtual Screening of Human O-GlcNAc Transferase Inhibitors
- Comparative Theoretical Studies on Several Energetic Substituted Dioxin-imidazole Derivatives
- Controlled Synthesis of PCL/PVP Copolymer by RAFT Method and Its Hydrophilic Block-Dependent Micellar Behaviors
- Epitaxial Growth and Thermoelectric Measurement of Bi2Te3/Sb Superlattice Nanowires
- Morphology and Growth Process of Bat-like ZnO Crystals by Thermal Evaporation
- Investigation of Ultrafast Electronic Transfer Process on Organic/Inorganic Heterojunction by Femtosecond Transient Absorption