TRAF3抑制NF-κB引起的肾囊腔形成作用*
2016-07-05孙丽萍胡巢凤张欣洲
孙丽萍, 胡巢凤, 张欣洲△
(1暨南大学第二临床医学院深圳市人民医院肾内科,广东 深圳 518020;2暨南大学医学院病理生理学教研室,广东 广州 510632)
TRAF3抑制NF-κB引起的肾囊腔形成作用*
孙丽萍1,胡巢凤2,张欣洲1△
(1暨南大学第二临床医学院深圳市人民医院肾内科,广东 深圳 518020;2暨南大学医学院病理生理学教研室,广东 广州 510632)
[摘要]目的: 研究肿瘤坏死因子受体相关因子3(TRAF3)对多囊肾集合管上皮细胞中核因子κB(NF-κB)信号通路及其下游产物表达的影响;观察TRAF3基因过表达的多囊肾集合管上皮细胞形成管状分支结构的变化,探讨TRAF3在多囊肾囊腔形成的发生发展中可能产生的作用。方法: Western blot检测多囊肾集合管上皮细胞和TRAF3基因过表达多囊肾集合管上皮细胞中NF-κB信号通路的变化,同时检测下游凋亡因子Bax 及 Bid 的表达及caspase-3 活性变化;通过Annexin V-FITC/PI双染法检测各组细胞的凋亡情况;应用三维(3D)培养法观察多囊肾集合管上皮细胞形成管状分支结构变化的差异。结果: TRAF3的过表达能显著抑制多囊肾集合管上皮细胞中 NF-κB 信号通路的活性,使下游的Bax及Bid表达显著下调,细胞凋亡明显减少;TRAF3 基因过表达多囊肾集合管上皮细胞管状分支结构较多囊肾集合管上皮细胞组明显增多。结论: TRAF3可能通过调节NF-κB 信号通路降低Bax及Bid活性,抑制细胞凋亡,从而抑制多囊肾囊腔形成。
[关键词]肿瘤坏死因子受体相关因子3; 核因子κB; 细胞凋亡; 囊腔形成,肾
多囊肾(polycystic kidney disease,PKD)是一种常见的常染色体遗传病,为单基因遗传,遗传学上将其分为显性和隐性2大类,两种类型均造成双侧肾脏发生病变。其中常染色体显性遗传性多囊肾病(autosomal dominant polycystic kidney disease,ADPKD)最常见,主要特点为肾小管上皮细胞来源的充满液体的囊泡不断地形成和扩张,且典型病例在中年时期会发展到终末期肾脏疾病(end-stage renal disease,ESRD)阶段,5%~10%使用肾脏替代治疗的 终末期肾脏病患者均由 ADPKD 引起[1]。常染色体隐性遗传性多囊肾病(autosomal recessive polycystic kidney disease,ARPKD)是一种多发于婴幼儿的遗传性疾病,全球发病率约为1/20 000[2]。ADPKD 多于成年后发病,发病率高(约为 1/400~1/1 000)且预后不良,目前临床上尚无有效治疗方法。因此,深入了解多囊肾病的发病机理,揭示其发生发展中的分子机制,针对致病基因发病机理进行干预,并探讨有效的早期诊断标志物及药物治疗靶点,具有重要意义。
肿瘤坏死因子受体相关因子(tumor necrosis factor receptor-associated factor,TRAF)家族是一类重要的胞内接头蛋白,能直接或间接与胞浆内的其它信号分子结合,从而参与多种下游信号通路的信号转导,进而调节细胞增殖、存活、凋亡[3]。目前已发现7种不同的TRAF蛋白,TRAF3就是其中的一员。TRAF3广泛存在于体内的各个组织,是核因子-κB(nuclear factor-κB,NF-κB)非经典通路重要的衔接蛋白。有研究发现[4-5],TRAF3表达下调可抑制 肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)引起的 NF-κB p100 的聚集并激活IKK复合物,引起NIK表达增加。我们前期研究发现[6-7],在多囊肾肝病1(polycystic kidney and hepatic disease 1,Pkhd1)基因部分沉默的内髓集合管细胞 (inner medullary collecting duct, IMCD)中,NF-κB处于持续性激活状态,并可导致细胞凋亡增加,并发现NF-κB促进三维(3D)培养的肾小管管腔增大,囊腔形成。因此,TRAF3 可能通过调节NF-κB通路,参与ARPKD肾集合管上皮细胞抗凋亡作用。本研究旨在探讨多囊肾细胞中 TRAF3 是否可以通过NF-κB信号通路抑制多囊肾囊腔形成。
材料和方法
1材料
1.1主要材料与试剂RPMI-1640 培养基、LHC-9 培养基、胎牛血清和青霉素链霉素双抗溶液(HyClone);EDTA-胰蛋白酶(Gibco);DMSO (Sigma);DBA 免疫组化试剂盒(ZYMED Laboratories Inc.);BCA 蛋白浓度测定试剂盒(Pierce);NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (碧云天生物技术研究所);Caspase-3 Activity Assay Kits(Calbiochem);兔抗鼠 NF-κB (p65) 抗体试剂盒、HRP标记的羊抗兔II抗(Cell Signaling Technology);兔抗鼠TRAF3多克隆抗体、兔抗鼠Bax单克隆抗体、兔抗鼠及Bid单克隆抗体(Santa Cruz);小鼠内髓集合管细胞株购自中国科学院细胞库;其它试剂均为 Fisher 产品。
1.2多囊肾小鼠模型的建立Pkhd1 基因第 15 和 16 外显子敲除的C57BL/6小鼠模型由南华大学贺修胜教授馈赠。由于Pkhd1 基因缺失,该小鼠模型的肝和肾表现出不同程度的管道扩张、囊肿和纤维化,有部分小鼠在胚胎即死亡,绝大部分小鼠由于肝肾囊肿而仅能存活 1 年;除此之外,Pkhd1-/-小鼠的胰腺和脑组织也出现囊肿和小管扩张;这些症状很好地模拟了人类 ARPKD。文中我们将这一Pkhd1 基因突变小鼠模型称为多囊肾小鼠。
1.3多囊肾集合管上皮细胞的分离及扩增培养将8周龄多囊肾小鼠乙醚麻醉,75%乙醇浸泡10 min;无菌条件下,打开腹腔,迅速取出肾脏,置于10 mL预冷的PBS(含5 mmol/L蔗糖)中,冰浴。用刀片切碎肾脏。用10 mL 0.5%胶原酶将其转移至10 mL离心管中,用吸管吹散混匀;在37 ℃条件下消化40 min,将消化后的组织液用70 μm孔径滤筛网过滤去除未被消化的结缔组织;过滤后室温1 000 r/min离心10 min。弃去上清后加入10 mL的PBS(含5 mmol/L蔗糖),混匀,室温1 000 r/min离心5 min,重复用PBS洗涤1次。弃去上清,加入含DBA抗体(终浓度10 μmol/L)的PBS(含5 mmol/L蔗糖)10 mL,4 ℃反应1 h,1 000 r/min离心5 min,用PBS(含5 mmol/L蔗糖)洗涤2次,完全弃去上清。将含有磁珠的1 mL PBS(含5 mmol/L蔗糖,可结合的最大细胞数为2×107)重悬后,转移至已弃去上清的含有细胞沉淀的离心管中,充分混匀,4 ℃反应30 min。置于磁柱上吸附5 min,吸掉液体,加入5 mL PBS(含5 mmol/L蔗糖),小心混匀,洗涤2次。弃去上清,加入含5 μL release buffer的无血清RPMI-1640 培养基 400 μL,混匀后37 ℃孵育20 min。置于磁柱上吸附5 min,将上清转移至15 mL离心管中,加入RPMI-1640 培养基洗涤2次,室温 1 000 r/min 离心 5 min。弃去上清,加入适量含5%胎牛血清的RPMI-1640 培养基混匀,加入24 孔板中(24孔板事先加入0.1%明胶包被,室温放置 30 min 以上),37 ℃培养箱中培养 48 h。待细胞贴壁后,将培养基换成含 γ-干扰素 的 LHC-9 培养基,33 ℃继续培养。所有实验用细胞都需转移至 37 ℃培养箱中培养 3 d后再进行实验研究。
2方法
2.1细胞培养小鼠多囊肾集合管上皮细胞和小鼠IMCD 细胞株均用含 10% 胎牛血清、1% 双抗的 RPMI-1640 培养基进行培养。3~4 d 细胞长到 90%左右融合时,PBS 缓冲液清洗,用胰酶消化,离心收集细胞,细胞计数仪计数后细胞按每孔 1×106个接种于 6 孔板。IMCD细胞是内髓集合管细胞,我们前期利用 RNAi 特异性抑制 Pkhd1 在 IMCD 细胞内的表达后发现,NF-κB处于持续性激活状态,并可导致细胞凋亡增加, IMCD 细胞不能在 3D培养条件形成管状分支结构,文中用此细胞作为从多囊肾小鼠肾脏提取的细胞的对照细胞。
2.2TRAF3基因转染构建pCDNA3.0-hTRAF3质粒,用 pCDNA 空载体质粒作为对照质粒,用 Lipofectamine 2000 (Invitrogen) 对细胞进行转染,按照试剂盒说明书进行操作。提取蛋白 用Western blot检测TRAF3的表达效率。
2.3细胞核蛋白提取按NE-PER Nuclear and Cytoplasmic Extraction Reagents Kit试剂盒说明书加入核蛋白抽提试剂提取细胞中核蛋白,终浓度调整为2 g/L。细胞培养至80%~90%融合,弃培养液,加3 mL预冷 PBS 漂洗细胞2次,弃漂洗液,取1.5 mL胞浆蛋白裂解液I漂洗细胞1次,弃漂洗液,取1.0 mL胞浆蛋白裂解液H于培养瓶中溶解细胞,吸净裂解液,转入1.5 mL离心管中,4 ℃、14 000 r/min离心2 min,分离上清,取0.5 mL胞浆蛋白裂解液I漂洗沉淀 1 次,4 ℃、14 000 r/inm离心2 min,取50 μL核裂解液重悬沉淀,4 ℃静置30 min,弃上清,4 ℃、14 000 r/min离心20 min,取上清液(即核抽提物)于-80 ℃保存。
2.4HE染色取材标本放置于 4%多聚甲醛中固定 18~24 h,脱水石蜡包埋,制成石蜡标本,4 μm 切片,每 1 例均连续切片,用于 HE 染色和免疫组织化学染色。组织片置于二甲苯中浸泡 15 min,更换二甲苯后再浸泡 2 min,在 100%、95%、90%、85%的乙醇中依次浸泡 3 min,流动自来水清洗 2 min,用苏木素染色 5 min,流动自来水冲洗 5 min,盐酸乙醇分化 2 min,自来水冲洗 5 min,70%乙醇浸泡 2 min,伊红染色 1 min,将载玻片依次放入 95%乙醇-100%乙醇×二甲苯II,各2 min,通风橱室温晾干,细胞核染成蓝色,细胞质为红色。光学显微镜观察、拍照。
2.5Western blot检测蛋白水平当细胞融合度为 80%~90%时,用RIPA裂解液冰上裂解 30 min,4 ℃、 12 000 r/min 离心 15 min,取上清液用BCA 法上酶标仪测定蛋白浓度;上样20 μg蛋白,加入等体积的2×SDS上样缓冲液,100 ℃加热5 min,使蛋白变性;30 mA 恒流行 7%~15% SDS-PAGE 分离样本,以 100 V 恒压转移到 PVDF 膜上,用 5% 脱脂奶粉溶液室温封闭1 h,分别加入兔抗鼠TRAF3(1∶800)、抗NF-κB(p65)、抗Bid(1∶1 000)及抗Bax抗体 (1∶1 000)室温孵育 2 h,次日用 TBST 漂洗 3次后加入 HRP 标记的II抗 (1∶5 000)室温孵育 1 h,TBST 清洗3 次加入 ECL 显色;通过凝胶成像系统扫描分析各蛋白变化。以β-actin 作为内参照。
2.6细胞免疫荧光染色4%多聚甲醛固定细胞,置于 4 ℃ 30 min;PBS 洗3遍后,用1% BSA+0.3% Triton X-100 室温透化 1 h。1 % BSA 溶液室温封闭2 h,加入适量 I 抗稀释液(1 % BSA 溶液)配制 I 抗,4 ℃反应过夜。PBS洗3次,加入Cy2/Cy3 标记 II 抗(1∶1 000 稀释于 1% BSA/PBS 中),避光室温反应 1 h,PBS 洗3 次,加入DAPI (1∶10 000 稀释于 PBS 中)室温反应 30 s;PBS 洗3 次。用封片剂 Aqueous Mountant 封片,盖玻片周围用指甲油密封。封片后荧光显微镜下进行观察或保存于 4 ℃后续观察。
2.7流式细胞术检测细胞凋亡取对数生长期的各组细胞,调节细胞浓度为1×108/L,接种于6孔培养板内;细胞长至80%融合时,无血清饥饿24 h诱导细胞凋亡;收集细胞,PBS缓冲液洗涤;重悬细胞于0.5 mL binding buffer中,加入2 μL Annexin V-EGFP 和5 μL 碘化丙啶(propidium iodide, PI),混匀后室温避光反应10 min,应用Single histogram statistic分析软件测定细胞凋亡率。
2.8Caspase-3的活性检测按照Calbiochem的Caspase-3 Activity Assay Kits 操作说明,离心收集细胞,使用预冷的PBS洗涤细胞2次,加裂解缓冲液重悬细胞,冰上孵育20 min,4 ℃、12 000 r/min离心5 min,取上清进行蛋白含量测定;加入反应试剂,与caspase-3的底物(Nacetyl-Asp-Glu-Val-Asp-p-nitroanilide)在37 ℃孵育4 h; 405 nm处测定吸光度(A)值。
2.93D细胞培养待细胞长至90%融合时,0.25%胰酶消化细胞,并用无血清1×RPMl-1640培养基清洗1遍。按下列比例配置胶原基质胶:375 μL H2O,100 μL 200 mmol/L HEPES,10×RPMI-1640 100 μL,50 μL 74 g/L NaHCO3,375 μL rat type I collagen,25 μL Matrigen,1滴 NaOH(pH7.4),5 μL内含3×106细胞的细胞液,混匀。96孔板每孔加入 100 μL 混有细胞的胶原基质胶,实验设置3个复孔。剩余的细胞再加入等体积的胶原基质胶使细胞浓度减半,再按3个复孔种于96孔板。置 37 ℃培养箱培养1h,待胶凝固后加入100 μL 培养基 (10%胎牛血清+1×RPMI-1640)。37 ℃培养箱培养 7 d后,显微镜下观察。
3统计学处理
所有实验均至少重复3次,各实验组数据采用SPSS 16.0统计软件处理。数据用均数±标准差(mean±SD)表示,组间比较用单因素方差分析,并用 SNK-q检验进行各组之间的两两比较,以P<0.05 为差异有统计学意义。
结果
1常染色体隐性遗传性多囊肾小鼠模型的鉴定
为了确定多囊肾小鼠模型作为肾囊肿研究的可行性,本实验收取从胚胎时期到出生后不同时期小鼠肾脏(胚胎15.5 d、18.5 d,出生后5 d、10 d),HE染色观察从胚胎时期到小鼠死亡不同时期肾脏囊性结构发展过程。结果发现多囊肾小鼠肾脏囊肿在胚胎15.5 d时已经出现囊性结构,随着时间的延长,胚胎18.5 d时,在髓质区囊性结构变大;出生后5 d,肾脏囊性结构进一步发生融合,变大;出生后10 d,肾脏大部分区域发生囊性化,皮质变薄,可见融合的囊性结构,见图1。
2多囊肾集合管上皮细胞中NF-κB的表达
从8周龄多囊肾小鼠中取出肾脏,通过应用双花扁豆凝集素(Dolichus biflorus agglutinin,DBA)分离方法,获得的多囊肾集合管上皮细胞系。DBA作为集合管细胞标志物[8],并用E-cadherin作为上皮细胞标准物[9],以鉴定细胞原始来源。免疫荧光染色显示本实验原代分离的细胞表达集合管细胞标志物DBA和上皮细胞标准物 E-cadherin。结果表明,我们已成功建立多囊肾集合管上皮细胞,见图2。
Figure 1.The changes of renal cysts in a mouse model of ARPKD in different periods under microscope.E:embryonic;P:postnatal (×10).
图1不同时期10倍镜下多囊肾小鼠肾脏囊肿的变化
Figure 2.The characteristics of collecting duct cell lines derived from the kidneys of mouse model of ARPKD. The cell lines were characterized by staining with collecting duct cell marker Dolichos biflorus agglutinin and epithelial cell markers E-cadherin. The scale bar=5 μm.
图2多囊肾小鼠肾脏集合管上皮细胞的特征
细胞鉴定成功后,在培养的细胞中加入TNF-α(质量浓度 50 μg/L)分别作用15 min和30 min后收取蛋白,Western blot检测细胞核NF-κB (p65)蛋白的表达情况,结果显示刺激15 min 时点多囊肾集合管上皮细胞组胞核 NF-κB (p65)蛋白表达水平明显高于IMCD组(P<0.05),30 min 时点胞核NF-κB(p65)表达水平与IMCD组比较也明显升高(P<0.05),且与15 min时点相当,见图 3。
3TRAF3 对 NF-κB 信号通路及Bax、Bid的作用
TRAF3基因过表达多囊肾集合管上皮细胞组(实验组)TRAF3表达显著增高。TNF-α 刺激15 min后,实验组细胞核NF-κB(p65)及细胞中Bax、Bid的表达较多囊肾集合管上皮细胞组(对照组)明显下降(P<0.05),而两组内Bax及Bid 的表达水平差异无统计学显著性。表明TRAF3 对NF-κB信号通路激活的抑制可以进一步影响下游凋亡因子的表达,见图4。
Figure 3.The expression of NF-κB (p65) in the ARPKD renal collecting duct epithelial cells. Mean±SD.n=3.*P<0.05vsIMCD group.
图3多囊肾集合管上皮细胞中NF-κB(p65)的表达
4TRAF3对细胞凋亡的影响
Annexin V-FITC/PI双染法检测细胞凋亡的实验结果显示,相对于IMCD组细胞,多囊肾集合管上皮细胞组中的细胞凋亡率显著增加(P<0.05);与多囊肾集合管上皮细胞组相比,过表达TRAF3的多囊肾集合管上皮细胞组细胞凋亡率明显减少(P<0.05)。
我们检测了caspase-3的酶活性,进而验证细胞凋亡中caspase-3活性的变化。结果发现,与IMCD组相比,多囊肾集合管上皮细胞组的caspase-3活性显著增加(P<0.05),而过表达TRAF3的多囊肾集合管上皮细胞组caspase-3活性则较多囊肾集合管上皮细胞显著降低。结果和Annexin V-FITC/PI双染的流式细胞术检测结果一致,见图5。
Figure 4.The activation of NF-κB signaling pathway in TRAF3 over-expressed cells. Control group was ARPKD renal collecting duct epithelial cells, experiment group was TRAF3 over-expressed ARPKD renal collecting duct epithelial cells. Mean±SD.n=3.*P<0.05vscontrol group.
图4TRAF3 过表达细胞中NF-κB信号通路激活情况
5TRAF3对3D培养条件下管状分支结构形成的影响
在胶原/基质胶三维培养条件下,80% IMCD细胞能形成肾内小管样分支结构;与此相反,约95%的多囊肾集合管上皮细胞管状分支的形成显著降低,而形成囊样结构,不能形成管状分支结构。接近60%的TRAF3过表达多囊肾集合管上皮细胞组能形成4个及其以上的分支,约 35%的细胞能形成1~3个分支,仅不足5%的细胞不能形成分支结构,见图6。
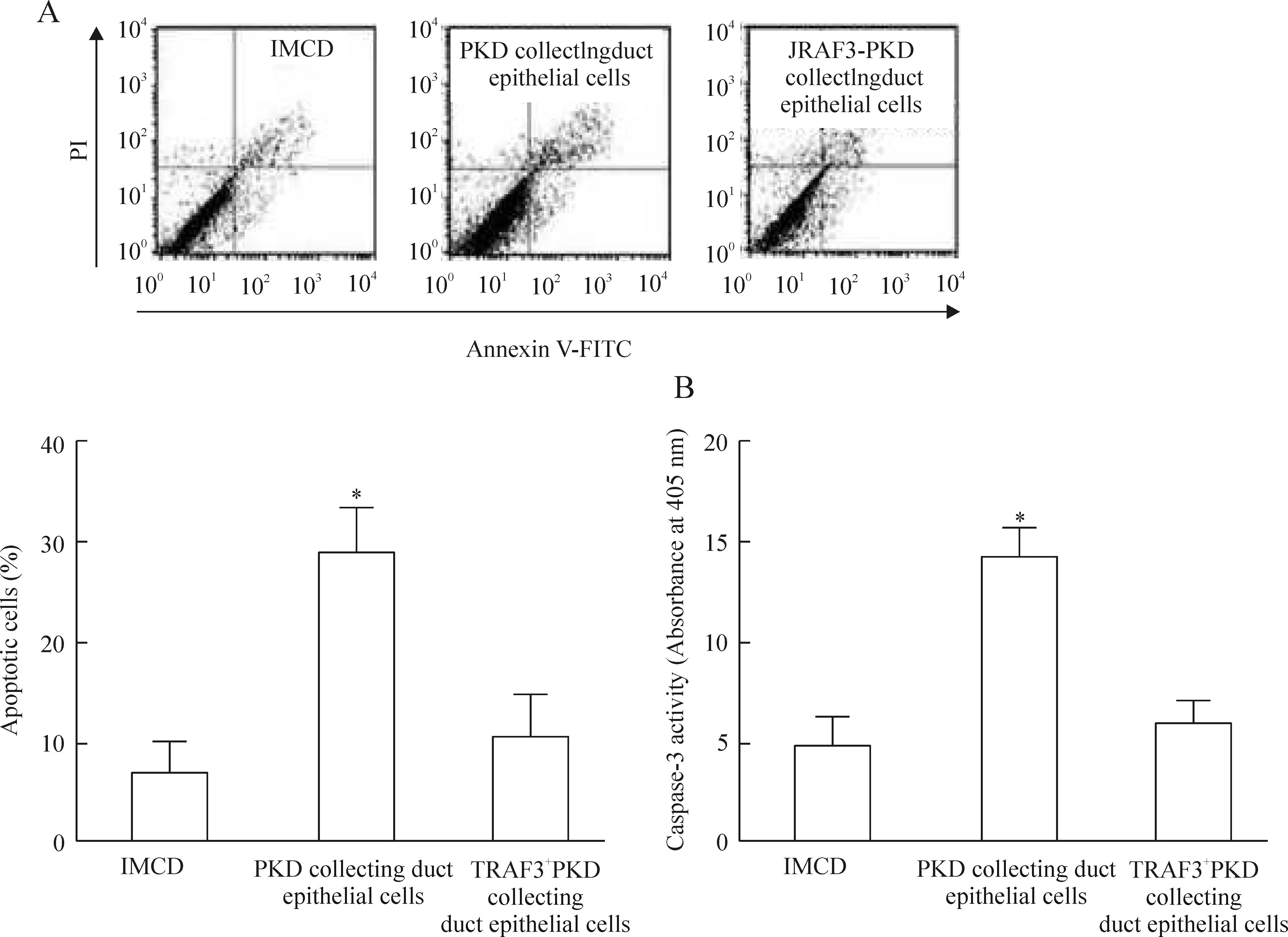
Figure 5.The apoptosis was analyzed in the cells of TRAF3 over-expressed ARPKD renal collecting duct epithelial cells. A: the apoptosis was determined by flow cytometry with Annexin V-FITC/PI double staining; B: the caspase-3 activity was measured. Mean±SD.n=3.*P<0.05vsIMCD and TRAF3+PKD collecting duct epithelial cells.
图5TRAF3过表达对细胞凋亡的影响
Figure 6.Tubulomorphogenesis was inducted in 3D culture of TRAF3 over-expressed PKD renal collecting duct epithelial cells. The scale bar=20 μm. A: IMCD cell; B: PKD renal collecting duct epithelial cell; C: TRAF3 over-expressed PKD renal collecting duct epithelial cell; D: cell tubulomorphogenesis counting. Mean±SD.n=3.*P<0.05vsIMCD group.
图6TRAF3过表达能促进多囊肾集合管上皮细胞形成管状分支结构
讨论
本研究通过对多囊肾集合管上皮细胞进行 TNF-α 刺激后检测 NF-κB 信号通路的变化,证实NF-κB信号通路在多囊肾中起一定作用,进一步检测过表达TRAF3 多囊肾集合管上皮细胞后 NF-κB 活性及其下游凋亡因子 Bax 及 Bid表达的变化,同时观察了细胞凋亡情况,并观察多囊肾集合管上皮细胞形成管状分支结构的变化。证实了过表达 TRAF3 可以下调下游凋亡因子的表达,抑制细胞凋亡,进而使多囊肾集合管上皮细胞形成管状分支结构。
多囊肾的发生是由人类肾脏小管生长发育分子失调所造成,因此揭示多囊肾发病机制,将有助于揭示肾脏小管发生、发育的分子机制[10]。我们前期研究证实了NF-κB 在FPC 缺陷细胞中处于持续性激活状态。而最近研究发现抑制TRAF3的表达引起经典和非经典NF-κB的共同激活。因此我们推测TRAF3可能通过调节NF-κB信号通路抑制下游凋亡因子的活性,从而参与肾囊肿形成过程。本研究将TRAF3基因转染多囊肾集合管上皮细胞使其过表达,发现相对于转染空载体质粒的对照组,TNF-α激活的 NF-κB通路中的Bax及Bid的表达水平明显下降,证明过表达TRAF3之后,多囊肾集合管上皮细胞中凋亡通路的激活受到抑制,使细胞凋亡减少。本研究结果显示:过表达TRAF3 细胞中TNF-α刺激的下游抗凋亡因子Bax及Bid表达量显著低于对照组,由此可知,TRAF3 在多囊肾集合管上皮细胞中可抑制NF-κB通路以及下游凋亡因子的激活与表达。
多项研究表明,小鼠肾集合管上皮细胞,在胶原/基质胶三维培养条件下能形成肾内小管样分支结构[11]。ADPKD 的致病基因(PKD1 和PKD2)缺失能抑制在三维培养条件下形成管状分支结构[12];而高表达PKD1能促进MDCK细胞管状分支结构形成[13];我们前期利用 RNAi 特异性抑制Pkhd1在IMCD细胞内的表达后发现,IMCD3 细胞不能在 3D培养条件形成管状分支结构。本研究发现,体外培养的TRAF3过表达多囊肾集合管上皮细胞组形成管状分支结构较多囊肾集合管上皮细胞管状分支的形成明显增多。证明 TRAF3 过表达多囊肾集合管上皮细胞中,囊腔形成明显减少。
综上所述,在多囊肾集合管上皮细胞中,TRAF3可显著抑制NF-κB信号通路以及下游凋亡因子表达。作为NF-κB 信号通路的一个负调控因子,TRAF3对于NF-κB引起的囊腔扩大具有抑制作用,可能成为多囊肾病中预防与保护肾小管结构破坏的潜在靶点。
[参考文献]
[1]Liu ZH. Nephrology in China[J]. Nat Rev Nephrol, 2013, 9(9):523-528.
[2]Halvorson CR, Bremmer MS, Jacobs SC. Polycystic kidney disease: inheritance, pathophysiology, prognosis, and treatment[J]. Int J Neph Renovas Dis, 2010, 3:69-83.
[3]Fick A, Lang I, Schafer V, et al. Studies of binding of tumor necrosis factor (TNF) -like weak inducer of apoptosis (TWEAK) to fibroblast growth factor inducible 14 (Fn14) [J]. J Biol Chem, 2012, 287(1):484-495.
[4]Hu H, Brittain GC, Chang JH, et al. OTUD7B controlls non-canonical NF-kappa B activation through deubiquitination of TRAF3[J]. Nature, 2013, 494 (7437):371-374.
[5]Yao Z, Xing L, Boyce BF. NF-kappa B p100 limits TNF-induced bone resorption in mice by a TRAF3-dependent mechanism[J]. J Clin Invest, 2009, 119(10): 3024-3034.
[6]Sun LP, Wang SX, Hu CF, et al. Down-regulation of PKHD1 induces cell apoptosis through PI3K and NF-κB pathways[J]. Exp Cell Res, 2011, 317(7): 932-940.
[7]Sun LP, Hu CF, Wang SX, et al. Constitutive activation of NF-κB causes tubule enlargement and cyst formation in cultured mammalian collecting duct cells[J]. Biotechnology (Indian), 2014, 10(21):13251-13256.
[8]Michael L, Sweeney DE, Davies JA. The lectin Dolichos biflorus agglutinin is a sensitive indicator of branching morphogenetic activity in the developing mouse metanephric collecting duct system[J]. J Anat, 2007, 210(1):89-97.
[9]Grande MT, Lopez-Novoa JM. Fibroblast activation and myofibroblast generation in obstructive nephropathy[J]. Nat Rev Nephrol, 2009, 5(6):319-328.
[10]孙丽萍,张欣洲,胡巢凤.Klotho:与FPC蛋白相互作用的蛋白分子[J]. 中国病理生理杂志, 2010, 26(3):518-523.
[11]Chen D, Roberts R, Pohl M, et al. Differential expression of collagen and laminin- binding integrins mediates ureteric bud and inner medullary collecting duct cell tubulogenesis[J]. Am J Physiol Renal Physiol, 2004, 287(4):F602-F611.
[12]Kim I, Ding T, Fu Y, et al. Conditional mutation of Pkd2 causes cystogenesis and upregulates beta-catenin[J]. J Am Soc Nephrol, 2009, 20(12):2556-2569.
[13]Boletta A, Qian F, Onuchic LF, et al. Polycystin-1, the gene product of PKD1, induces resistance to apoptosis and spontaneous tubulogenesis in MDCK cells[J]. Mol Cell, 2000, 6(5):267-273.
(责任编辑: 卢萍, 余小慧)
Effects of TRAF3 on inhibiting cyst formation induced by NF-κB signaling in kidney
SUN Li-ping1, HU Chao-feng2, ZHANG Xin-zhou1
(1DepartmentofNephrology,ShenzhenPeople’sHospital,SecondClinicalMedicalCollege,JinanUniversity,Shenzhen518020,China;2DepartmentofPathophysiology,SchoolofMedicine,JinanUniversity,Guangzhou510632,China.E-mail:xin.zhou@medmail.com.cn)
[KEY WORDS]Tumor necrosis factor receptor associated factor 3; NF-κB; Apoptosis; Cyst formation, kidney
[ABSTRACT]AIM: To investigate the effects of tumor necrosis factor (TNF) receptor associated factor 3 (TRAF3) on the signaling pathway of NF-κB and the expression of Bax and Bid in renal collecting duct epithelial cells of polycystic kidney disease (PKD), and to observe the tubulogenesis of TRAF3 transgenics for exploring the protective effect of TRAF3 on the cystogenesis and development of PKD. METHODS: The signaling changes of NF-κB and expression of Bax and Bid in PKD renal collecting duct epithelial cells and TRAF3 transgenic PKD renal collecting duct epithelial cells were determined by the Western blot. The percentage of apoptotic cells was measured by flow cytometry with Annexin V staining. The caspase-3 activity was also measured. The 3D Matrigel culture was performed to examine abnormal tubulomorphogenesisinvitro. RESULTS: The over-expression of TRAF3 significantly inhibited the signaling pathway of NF-κB in PKD renal collecting duct epithelial cells and significantly downregulated the expression of Bax and Bid, and also decreased the cell apoptosis. More branch structures were observed in the TRAF3 transgenic PKD renal collecting duct epithelial cells. CONCLUSION: TRAF3 is a negative regulatory inhibitor for Bax and Bid by regulating the NF-κB signaling and may be a new target for inhibiting the cyst formation in PKD.
[文章编号]1000- 4718(2016)06- 1077- 07
[收稿日期]2016- 03- 31[修回日期] 2016- 04- 29
*[基金项目]广东省科技计划(No. 2014A020212472);深圳市科技计划(No. JCYJ20150403101146288)
通讯作者△Tel: 0755-22943522; E-mail: xin.zhou@medmail.com.cn
[中图分类号]R681.3; R363.2
[文献标志码]A
doi:10.3969/j.issn.1000- 4718.2016.06.020
