3M综合征家系报道及临床分析
2016-06-18张亚男皮亚雷李玉倩齐占江张会丰
张亚男,皮亚雷,阎 雪,李玉倩,齐占江,张会丰
(河北医科大学第二医院儿科,河北 石家庄 050000)
·研究快报·
3M综合征家系报道及临床分析
张亚男,皮亚雷,阎 雪*,李玉倩,齐占江,张会丰
(河北医科大学第二医院儿科,河北 石家庄 050000)
3M综合征;胎儿生长迟缓;诊断
1975年Miller等[1]首次报道了3M综合征(OMIM#273750)这一罕见的常染色体隐性遗传病,其主要临床表现为严重宫内和出生后生长迟缓。患儿出生后主要表现为身材矮小,面部畸形,管状骨细长和脊椎骨椎体较高,不伴有智力异常和其他脏器的损害。致病基因包括CUL7(Cullin7)、OBSL1(Obscurin-Like 1)和CCDC8[2](Coiled-coil domain containing 8,CCDC8,也称为P90),其中CUL7基因突变导致的3M综合征占84%[3]。迄今国内未见此类病例报道。本研究初步探讨了一个家系中3例3M综合征患儿的临床特征及致病基因特点,报告如下。
1 资料与方法
1.1 一般资料 首诊患儿,女性,14岁,为先证者,河北省邢台市人,就诊原因是身高落后于同龄儿童;患儿足月顺产,其母孕期无异常,生后无窒息,出生体质量1.5 kg,出生身长较小(具体不详)。自幼生长缓慢,生长速度为3~5 cm/年,头围较大,与身高不成比例,伴有面部畸形,鼻梁扁平,鼻孔向上,嘴唇厚,下颌宽。10岁乳房发育,12岁月经初潮,无明显身高突增。无智力运动发育异常。就诊时身高130.0 cm(-5.0 SD),体质量36 kg。父母非近亲结婚,父亲身高170.2 cm,母亲身高158.0 cm。患儿有同胞妹妹1个,同胞弟弟1个,均有生长迟缓。患儿妹妹,10岁,身高110.0 cm(-4.816 SD),双侧乳房开始发育。患儿弟弟,9岁,身高105.8 cm(-5.1SD),体质量18 kg,尚未出现第二性征发育。
1.2 辅助检查 先证者:肝功能正常;甲状腺功能正常;骨龄15岁。妇科盆腔超声显示:子宫前位,大小约3.78 cm×4.14 cm×3.48 cm,子宫内膜厚度约1.29 cm;左侧卵巢2.61 cm×1.47 cm×1.8 cm,右侧卵巢3.46 cm×2.29 cm×2.34 cm,内可见黄体回声。
先证者胞弟:肝肾功能正常;生长激素激发试验显示生长激素峰值为17.2 μg/L;胰岛素样生长因子1(insulin-like growth factor 1,IGF-1)为740 ng/L;甲状腺功能正常;垂体MRI显示未见异常。
先证者胞妹:拒绝接受检查。
1.3 方法
1.3.1 基因芯片捕获及高通量测序 取患者全血2 mL,用DNA Extraction kit按照说明提取外周血淋巴细胞DNA,并使用Nanodrop 2000将DNA随机打断成350~450 bp的随机片段。DNA的制备按照Illumina 标准化流程进行。 用GenCap custom enrichment kit(Mygenostics,Beijing)对144个与骨骼发育相关基因的外显子及其侧翼序列进行捕获,制备成DNA文库。提取先证者的DNA,与GenCap probe(Mygenostics,Beijing)混合,PCR扩增,然后进行杂交。DNA库在Illumina HiSeq 2000测序平台上使用高保真DNA聚合酶进行扩增并进行高通量测序。应用SOAPsnp程序进行单核苷酸序列多态性分析;用GATK软件进行插入缺失分析;最后对生物多态性的信息进行收集,并通过应用Four algorithms、PolyPhen、SIFT、Mutationtaster 和 PMut预测其致病性。
1.3.2 Sanger法验证 对发现的致病突变在其所在片段上、下游设计引物进行PCR扩增,对产物作Sanger测序,从而验证基因芯片捕获和高通量测序的结果。对患者父母也采用Sanger法进行目标基因突变验证。
以上检测过程由北京麦基诺临床检验中心完成。本研究获得河北医科大学第二医院医学伦理委员会的审批并经患儿家属的知情同意。
2 结 果
2.1 临床资料 患者主要表现为宫内和生后生长迟缓,身材矮小,头围与身长不成比例,伴有面部畸形,鼻梁扁平,鼻孔向上,嘴唇厚,下颌宽。生长激素激发试验和IGF-1水平正常。甲状腺功能正常。智力发育和第二性征发育正常。
2.2 基因分析 先证者的CUL7基因第15外显子发现了1个c.3232 C>T(p.Q1078X)终止突变和第4外显子发现c.1219-1238缺失导致的移码突变(p.S407fs),这2个基因突变分别来自父亲和母亲。这2个基因位点的突变未见临床致病性报道。另外,对先证者的妹妹和弟弟也进行了基因分析,发现其妹妹、弟弟的基因变异类型与先证者相同。见图1。
2.3 家系患病情况 先证者的父母均健康。先证者的妹妹和弟弟均患病。家系图见图2。

图1 基因分析
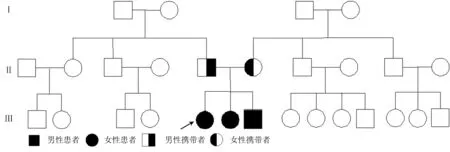
图2 家系患病情况
3 讨 论
3M综合征是一种罕见的骨骼发育异常疾病,为常染色体隐性遗传,其特征是宫内发育迟缓和严重的生后生长迟缓,表现为四肢管状骨细长和椎体较高,常常还伴有面部畸形,包括三角脸、前额突出、鼻梁扁平、鼻孔向上、嘴唇丰满、下颌宽,但是患儿智力正常,且没有其他重要脏器受累[1-3]。
该家族的3例同胞姐弟均表现为宫内和生后生长迟缓,年生长速度<5 cm,头围较大,与身高不成比例,两侧肢体对称,无腰椎前突,智力发育正常。先证者及其胞弟甲状腺功能均正常,IGF-1也在正常范围,左手正位片显示管状骨细长。垂体的MRI正常。先证者胞弟生长激素激发试验的生长激素峰值高达17.2 μg/L。Hanson等[4]研究显示,大部分3M综合征患儿生长激素激发试验的峰值正常,IGF-1处于正常或者偏低的水平,而胰岛素样生长因子结合蛋白,特别是胰岛素样生长因子结合蛋白3的水平显著降低。先证者胞弟生长激素激发试验正常,IGF-1水平正常。Hanson等[4]通过对3M综合征患儿皮肤成纤维细胞株研究发现,CUL7-/-细胞IGF-1信号传导途径、CCDC8-/-细胞生长激素信号传导途径、OBSL1-/-细胞的生长激素信号传导途径和IGF-1信号传导途径均受损。提示3M综合征患儿不存在生长激素分泌异常,预示生长激素治疗效果欠佳。
先证者已有月经初潮且月经规律,子宫和卵巢的超声均正常。先证者妹妹,10岁,已开始乳房发育。先证者弟弟,9岁,未出现第二性征发育。表明3M综合征患儿青春期启动和过程正常。Meazza等[5]对1例男性3M综合征患者进行了18年的追踪调查,发现患者有正常的青春期发育,最终睾丸的容积能达到正常人成年男性水平,性激素水平也正常。
本研究的3例3M综合征患儿基因检测结果显示,CUL7基因第15外显子发现了1个c.3232 C>T终止突变(p.Q1078X)和第4外显子发现c.1219-1238缺失导致的移码突变(p.S407fs)。这2个基因突变分别来自父亲和母亲。CUL7基因位于在6p21.1 区,含有26个外显子,编码形成含有1 698个氨基酸的CUL7蛋白[6]。CUL7是Cullin家族中的一员,它与SKP1-FBX29(FBXW8)和ROC1构成泛素连接酶E3,其中CUL7起骨架作用。CUL7基因的无义或者错义突变使CUL7蛋白不能招募ROC1,从而使底物不能泛素化,无法降解,在体内堆积[7]。CUL7基因敲除小鼠胚胎出现胰岛素受体底物1堆积,它的堆积导致下游底物Akt和MEK/ERK激活增加,从而导致生长迟缓[8]。另外,CUL7蛋白可以与p53结合,抑制p53的转录激活效应,从而促进细胞增殖[9]。而CUL7-/-小鼠出现宫内生长迟缓,其胚胎的成纤维细胞的p53的活性显著增加[10]。患3M综合征的胎儿胫骨生长板的组织形态学表现为:静止带和增殖带的软骨细胞的体积增大、密度增加;细胞外基质的合成受损;前肥大细胞带和肥大细胞带未见明显异常。表明CUL7参与了软骨细胞的生长和增殖[11]。
CUL7基因突变导致了3M综合征的发病,为探求儿童生长迟缓的病因提供了新的思路。另外,对于存在宫内和生后生长迟缓、头围与身高不成比例的身材矮小患儿,应进行基因的检测,明确致病基因和遗传方式,对指导优生优育有重要意义。
[1] Miller JD, McKusick VA, Malvaux P, et al.The 3M syndrome:a heritable low birthweight dwarfism[J]. Birth Defects Orig Artic Ser,1975,11(5):39-47.
[2] Harson D,Steven A,Phillip GM,et al.Identifying biological pathway that underlie primordial short stature using network analysis[J]. J Mol Endocrinol,2014,52(3):333-334.
[3] Huber C, Dias-Santagata D, Glaser A,et al. Identification of mutations in CUL7 in 3-M syndrome[J]. Nat Genet,2005,37:1119-1124.
[4] Harson D,Murray PG,Coulson T,et al.Mutation in CUL7,OBSL1 and CCDC8 in 3-M syndrome lead to disordered growth factor signaling[J]. J Mol Endocrinol,2012,49(3):267-275.
[5] Meazza C,Lausch E, Pagani S,et al. 3-M syndrome associated with growth hormone deficiency:18 year follow-up of a patient[J]. Ital J Pediatr,2013,39:21.
[6] Dias DC, Dolios G,Wang R,et al. CUL7:A DOC domain-containing cullin selectively binds Skp1.Fbx29 to form an SCF-like complex[J]. Proc Natl Acad Sci USA,2002,99(26):16601-16606.
[7] Huber C, Dias-Santagata D, Glaser A et al.Identification of mutations in CUL7 in 3-M syndrome[J]. Nat Genet,2005,37(10):1119-1124.
[8] Xu X,Sarikas A,Dias-Santagata DC,et al. The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation[J]. Mol Cell,2008,30(4):403-414
[9] Andrews P, He YJ, Xiong Y. Cytoplasmic localized ubiquitin ligase cullin 7 binds to p53 and promotes cell growth by antagonizingp53 function[J]. Oncogene,2006,25(33):4534-4548.
[10] Arai T, Kasper JS, Skaar JR, et al. Targeted disruption of p185/Cul7 gene results in abnormal vascular morphogenesis[J]. Proc Natl Acad Sci USA,2003,100(17):9855-9860.
[11] Huber C,Delezoide AL,Guimiot F,et al.A large-scale mutation search reveals genetic heterogeneity in 3M syndrome[J]. Eur J Hum Genet,2009,17(3):395-400.
2016-03-03;
2016-03-23
张亚男(1984-),女,河北正定人,河北医科大学第二医院主治医师,医学硕士,从事儿科内分泌遗传代谢研究。
*通讯作者。E-mail:ywcyx752@163.com
10.3969/j.issn.1007-3205.2016.04.025
R596
B
1007-3205(2016)04-0464-03
刘斯静)
