Modulation by desensitized nicotinic receptors on metabolism of DA in striatum derived from the hemiparkinsonian model
2016-06-05FurongHANHaiWANG
Fu-rong HAN, Hai WANG
Modulation by desensitized nicotinic receptors on metabolism of DA in striatum derived from the hemiparkinsonian model
Fu-rong HAN1,2, Hai WANG2,3
1. Department of Pharmacy, Beijing Tongren Hospital, Capital Medicine University, Beijing 100730, China;
2. Cardiovascular Drug Research Center, Academy of Military Medical Sciences, Beijing 100850, China;
3. Cardiovascular Drug Research Center, Thadweik Academy of Medicine, Beijing 100039, China
Objecti ve:The purpose of this study was to investi gate the eff ects of desensiti zed nicoti nic receptors (nAChRs) on striatal dopaminergic system in the hemiparkinsonian rats treated with 6-hydroxydopamine (6-OHDA).Methods:We examined the eff ects of desensiti zed nAChRs on the levels of dopamine (DA) and its metabolites, mRNA expression of dopamine receptor D1,D2 and monoamine oxidase B (MAO-B) in the striatum of 6-OHDA-lesioned rats using high-performance liquid chromatography and reverse transcripti on-polymerase chain reacti on.Results:The results showed that nAChRs desensiti zati on following repeated nicoti ne sti mulati on could reverse signifi cantly the decrease of striatal DA and its metabolites levels and the increase in DA turnover in lesioned side striatum of hemiparkinsonian rats. Dopamine D1 receptor mRNA expression increased signifi cantly, whereas dopamine D2 receptor mRNA expression remained unchanged in lesioned side striatum of nicoti ne-treated rats compared to 6-OHDA-lesioned rats when nAChRs were desensiti zed. Meanwhile, nicoti ne-treated rats displayed a signifi cant decrease in MAO-B mRNA expression in lesioned side striatum compared to 6-OHDA-lesioned rats after nAChRs desensitization.Conclusion:These results suggest that nAChRs desensiti zati on could promote DA level, upregulate dopamine D1 receptor expression and downregulate MAO-B expression in striatum of hemiparkinsonian rats.
Parkinson’s disease; nicoti ne; nicoti ne acetylcholine receptor; desensiti zati on
Introduction
Parkinson’s disease (PD) is a common neurodegenerative disorder of unknown etiology, aff ecting about 1-3% of the population over 50 years of age and characterized by relatively selective nigrostriatal dopaminergic degeneration, with the subsequent loss of dopamine in the striatum. Th e resulting dopaminergic deficit at the striatal dopamine receptors causes the characteristic symptoms of bradykinesia, tremor, rigidity, and postural abnormalities[1]. Although replenishment of dopamine in the brain by administration of the dopamine precursor L-3,4- dihydroxyphenylalanine has shown certain usefulnessin the treatment of PD, this treatment is at best palliative and does not address the underlying pathophysiology, thereby can not halt or retard degeneration of dopaminergic neurons[2]. Th erefore, for the treatment of PD, it is desirable to develop other therapeutic interventions that improve dopaminergic function by mechanisms distinct from the current PD therapy. Accumulating data from epidemiological studies suggests that there is a negative correlation between cigarette smoking and the occurrence of PD[3-4]. Th is observation suggests that nicotine might have a neuroprotective eff ect on the dopaminergic neurons. Further studies showed that nicotine could reduce the degeneration of dopaminergic neurons in substantia nigra pars compacta (SNpc), promote dopamine synthesis and release, and increase extracellular dopamine levels in striatum[5-6]. Based on these fi ndings, nicotine derivatives may have a therapeutic application in the treatment of PD.
As a specific and highly selective ligand for nicotinic acetylcholine receptors (nAChRs), nicotine binds readily to nAChRs and exerts its function by acting on nAChRs. However, acute or chronic repetition of nicotine exposure during smoking can induce nAChRs desensitization, causing a decrease or loss of N-like response to agonists[7-9]. Traditionally, desensitized nAChR is viewed as a non-functional state. However, studies from our laboratory have shown that nAChRs desensitization only causes a decrease or loss of N-like response to agonists but is not a nonfunctional state. We proposed that desensitized nAChRs have specific functions[10]. We have demonstrated that desensitized nAChRs could enhance the functions of dopamine receptors[11]. In addition, we have found that when brain nAChRs were in a subacute, acute or chronic desensitized state, induced by repeated administration of nicotine, activities of cAMP-dependent protein kinase A (PKA) and protein kinase C (PKC) were decreased[12]. Furthermore, it has previously been documented that rats with hemiparkinsonism following unilateral 6-OHDA injection have a signifi cant increase in PKA and PKC activities[13-14]. These observations suggest a possible role for desensitized nAChRs in the regulation of the dopaminergic system. Therefore, we performed a detailed examination of changes in apomorphine-induced rotational behavior in rats with unilateral 6-OHDA-induced lesions when nAChRs were in diff erent desensitized states. We found that hemiparkinsonian rats displayed a significant reduction in apomorphine-induced rotational behavior when brain nAChRs were in diff erent desensitized states induced by repeated administration of nicotine[15]. To identify the detailed mechanism by which desensitized nAChRs causes this effect, the present study further observe the effects of desensitized nAChRs on the levels of dopamine and its metabolites and mRNA expression of dopamine receptor D1, D2and monoamine oxidase B (MAO-B) in the 6-OHDA rat model of PD.
Materials and Methods
Animals
Male Wistar rats, weighing 180–220 g, obtained from the laboratory animal center of Beijing Institute of Pharmacology and Toxicology, were used in this study. All animals were housed in a ventilated animal room under a light: dark cycle of 12:12 h (lights on from 06:00 to 18:00 h). Food and water were provided ad libitum. The study was conducted in accord with the principles and procedures of the NIH Guide for the Care and Use of Laboratory Animals.
6-OHDA lessions
6-OHDA solution (2 μg base per μl, Sigma, St. Louis, MO, USA) was prepared by dissolving 6-OHDA hydrochloride in ice-cold saline containing 0.02% ascorbic acid. Th e rats were anesthetized with sodium pentobarbital (40 mg/kg, i.p.) and immobilized in a stereotaxic frame. The 6-OHDA solution was injected into each rat’s left substantia nigra pars compacta using coordinates relative to the bregma and dura (A: –5.3 mm from bregma, L: +1.8 mm from midline and H: –7.8 mm from dura) according to the atlas of Paxinos and Watson[16]. A total of 8 μg of 6-OHDA base was infused at a fl ow rate of 1 μg/min. Upon completion, the injection cannula was kept in place for an additional 10 min to prevent backfl ow of the solution. Proper post-operative care was taken till the animals recovered completely. Rats were screened three weeks aft er surgery by monitoring rotational locomotor. Rotational behavior was assessed by placing rats in 50 cm truly hemispheric bowls, and rotations were counted aft er apomorphine administration (0.5 mg/kg, s.c., Sigma, St. Louis, MO, USA). Contralateral rotations induced by apomorphine were measured once a week for two weeks. Only those rats showing at least 210 turns during the 30-min test were selected for further study[17].
Nicotine treatment
The animals were randomly divided into three groups: control group, 6-OHDA-lesioned group and nicotine-treated lesioned group. Nicotine (Sigma, St. Louis, MO, USA) was administrated at 2.4 mg/kg/ day for 14 days, and then nAChRs enter a chronic desensitized state[18].
Histological examination
After animals were decapitated, the midbrains were removed and fi xed with 10% (vol/vol) buff ered formaldehyde. Tissue samples were dehydrated, cleared and embedded in paraffi n. Representative midbrain sections (7 μm thick) were deparaffinized with xylene, stained with hematoxylin and eosin (HE) and observed under a stereozoom microscope (Olympus, Japan) and photographed. HE-stained neurons of the SNpc were counted manually (Light microscopy; ×400).
High-performance liquid chromatography (HPLC) analysis
Animals were killed by cervical dislocation and brains were rapidly removed on a frozen block. Striata were carefully dissected using laboratory microscope, weighed, thereafter immediately frozen and stored at -70°C until further use. Dopamine and its metabolites concentrations were measured using HPLC with electrochemical detection (ECD) and glassy carbon electrode. Striata were homogenized in ice-cold 0.1 mol/L HCLO4solution and centrifuged 12 000 r/min for 20 min to precipitate proteins. Supernatant was removed, fi ltered and examined for its contents of dopamine (DA), dihydroxyphenyl acetic acid (DOPAC) and homovanillic acid (HVA) (standard substances supplied by Sigma, St. Louis, MO, USA). The supernatant (12 000 r/min for 20 min) was injected (50 μl) into the HPLC system (Agilent, USA) equipped with HP 1049A amperometric detector and C18, ion pair, analytical column (150×4.6 mm; Agilent, USA). The mobile phase contained 85 mM citric acid, 100 mmol/L sodium acetate, 0.2 mM Na2-EDTA, 0.9 mmol/L sodium octylsulfate, and 15% methanol. The mobile phase was adjusted to pH of 3.7 with 85% orthophosphoric acid and was degassed twice using a 0.45 Am membrane fi lter prior to use. Th e mobile phase fl ow rate was 1 ml/min, and the working electrode was kept at 0.70 V. Levels of DA, DOPAC and HVA were calculated from the comparison of simple peak area with external standard peak region from DA, DOPAC and HVA and were expressed as picomole per milligram (pmol/mg) of tissue.
Reverse transcription-polymerase chain reaction (RTPCR) analysis
Total RNA was isolated from striata with TRIzol reagent as described by the supplier (Invitrogen Canada Inc, Burlington, ON). Th e RNA product was resuspended in 20 μl diethyl pyrocarbonate-treated water. The quality of RNA samples was confirmed by electrophoresis of RNA through 1.5% agarose gel containing ethidium bromide and visualization by UV illumination. RNA was stored at -70 °C until use. Total RNA was reverse transcribed at 42 °C for 1 h with moloney murine leukemia virus (MMLV) reverse transcriptase according to the instruction of the manufacturer of the reagent (Promega, USA). Following the RT reaction, the cDNA products were stored at -20 °C until use. cDNA was amplifi ed using adequate primers. Dopamine D receptor primers were: forward 5’-ATCTCTTGGTGGCTGTCCTG-3’; reverse, 5’-GTTGTCATCCTCGGTGTCC T-3’. Dopamine D2receptor primers were: forward 5’-AAC CCGGACCTCCCTTAAGAC-3’; reverse, 5’-TTGCGGAACTCGATGTTGAAG-3’. MAO-B primers were: forward 5’-CCGAGACAGCTTCACATTGGA-3’; reverse, 5’-CCAGGAAACCAA GAGCTGTTG-3’. Negative control reaction, without template or MMLV reverse transcriptase, was included in PCR amplifi cation with primer set in parallel. As a control to eliminate variations for sample-to-sample diff erences in RNA extraction and conversion to cDNA, we amplifi ed the housekeeping gene β-actin. β-actin primers were: forward 5’-AGGCATCCTGACCCTGAAGTA C-3’; reverse, 5’-TCTTCATGAGGTAGTCTGTCA G-3’. Following a 15-min preincubation at 95 °C, the parameters were as follows: dopamine D1receptor, 32 cycles, 94 °C for 30 s, 62 °C for 30s, 72 °C for 1 min; dopamine D2receptor, 32 cycles, 94 °C for 30 s, 58 °C for 30 s, 72 °C for 1 min; MAO-B, 32 cycles, 94 °C for 30 s, 68 °C for 30 s, 72 °C for 1 min, a 10 min incubation at 72 °C followed each PCR reaction. PCR products were separated on 1.5% agarose gels stained with ethidium bromide, visualized under UV light and optical density data used for semiquantitation of mRNA expression.
Data analysis
Data are presented as mean ± standard error of the mean. Statistical differences between two groups were determined by one-way ANOVA followed by post hoc Neuman–Keuls test with the statistical signifi cance set at a P value of < 0.05.
Results
Modulation by desensitized nAChRs on dopamine and its metabolites
HPLC analysis revealed that unilateral 6-OHDA-lesioned rats exhibited a signifi cantly lower level of DA and its metabolites DOPAC and HVA in lesioned side of the striatum compared to control rats. When nAChRs were desensitized following chronic nicotine treatment, nicotine-treated rats exhibited a signifi cant increase in the levels of dopamine and its metabolites in lesioned side striatum compared to 6-OHDA-lesioned rats (Fig.1A).
The ratio of (DOPAC+HVA) to DA was used as an index of the rate of DA metabolism. This ratio was signifi cantly elevated in lesioned side striatum of 6-OHDA-lesioned rats in comparison with controlrats. When nAChRs were desensitized following repeated nicotine stimulation, the (DOPAC+HVA)/DA ratios in lesioned side striatum of nicotine-treated lesioned rats were signifi cantly decreased compared to 6-OHDA-lesioned rats(Fig.1B).
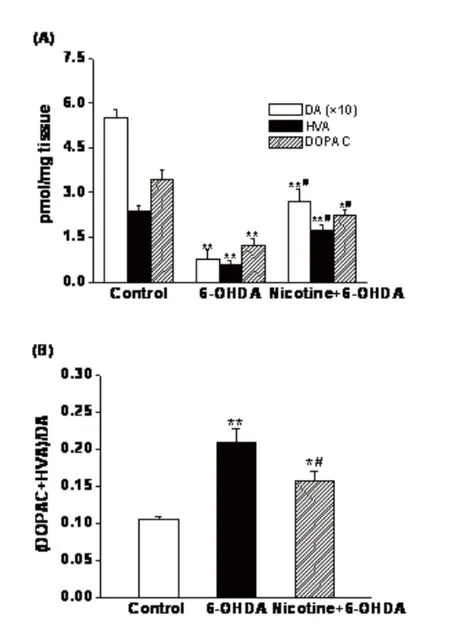
Fig.1 Effects of desensitized nicotinic receptors on 6-OHDA-leisioned rat striatal (A) dopamine and its metabolites levels and (B) dopamine turnover. Desensitization of nicotinic receptors was induced by repeated administration of nicotine at 2.4 mg/kg/day for 14 days. Dopamine and its metabolites in the striatum was measured with HPLC-ECD. Values are expressed as picomole per milligram (pmol/mg) of fresh striatum weight and given as mean ± SEM, n = 6,*P< 0.05,**P< 0.01 vs controls;#P<0.05 vs 6-OHDA-leisioned group.
Modulation by desensitized nAChRs on dopamine receptors and MAO-B mRNA expression
Semiquantitative RT-PCR analysis revealed that Dopamine D1receptor mRNA expression decreased signifi cantly in lesioned side striatum aft er 6-OHDA lesioning. However, nicotine-treated rats displayed a signifi cant increase in dopamine D1receptor mRNA expression in lesioned side striatum compared to 6-OHDA-lesioned rats when nAChRs were desensitized following chronic nicotine stimulation(Fig.2A). Unilateral 6-OHDA-lesioned rats exhibited a significant increase in dopamine D2receptor mRNA expression in the lesioned side striatum compared to control rats. When nAChRs were desensitized induce by repeated nicotine treatment, dopamine D2receptor mRNA expression remained unchanged in lesioned side striatum of nicotine-treated rats compared to 6-OHDA-lesiond group(Fig.2B).
MAO-B mRNA expression was determined in striatal samples, as shown in Figure 2. C Th ere were no signifi cant diff erences in MAO-B mRNA expression between control and lesioned rats. However, MAO-B mRNA expression decreased significantly in the lesioned side striatum of nicotine-treated rats compared to 6-OHDA-lesioned rats when nAChR is desensitized following repeated nicotine stimulation. Th ere was a signifi cant decrease in the amount of hematoxylin and eosin stained neurons in lesioned side SNpc region of 6-OHDA-lesioned rats compared to the SNpc of control rats. As shown in Figure 3A-D, in comparison with control rats, most of the neurons in the SNpc on the lesioned side were shrunken and some of them showed a loss of their nucleus. When nAChR is desensitized following repeated nicotine stimulation, there is no significant change in the number of hematoxylin and eosin stained neurons and neurons morphology in the lesioned SNpc region between 6-OHDA-lesioned and nicotine treated lesioned rats.
Discussion
In our previous study, we have examined the eff ect of desensitized nAChRs on the behavioral recovery in a rat 6-OHDA-induced PD model. Th e results showed that hemiparkinsonian rats displayed a significant reduction in apomorphine-induced rotational behavior when brain nAChRs were in subacute, acute or chronic desensitized states induced by repeated administration of nicotine[15]. Th is fi nding suggested that desensitized nAChRs can lead to behavioral improvement in the 6-OHDA rat model of PD and improve the decline in the striatal dopaminergic function of 6-OHDA-lesioned rats. To gain insight into the mechanism by which desensitized nAChRs causes this eff ect, the present study investigated the effects of desensitized nAChRs on the levels of dopamine and its metabolites and mRNA expression of dopamine receptor D1, D2and MAO-B in the 6-OHDA rat model of PD. Furthermore, we observed the pathological changes of SNpc region in 6-OHDA model of PD.

Fig. 2 Effects of desensitized nicotinic receptors on dopamine D1receptor(A), dopamine D2receptor(B) and monoamine oxidase B (MAO-B)(C) mRNA levels in the striatum of 6-OHDA-leisioned rats(A). Dopamine D1receptor, dopamine D2receptor and MAO-B mRNA expression was measured with semiquantitative RT-PCR. Desensitization of nicotinic receptors was induced by repeated administration of nicotine at 2.4 mg/kg/day for 14 days. Values are expressed as mean ± SEM, n = 6,*P< 0.05 vs controls;#P<0.05 vs 6-OHDA-leisioned group.
The results in this study showed that unilateral 6-OHDA-lesioned rats exhibited a significant decrease in the levels of dopamine and its metabolites and a concomitant increase in dopamine turnover in lesioned side of the striatum compared to the striatum of control rats. RT-PCR analysis revealed that dopamine D1receptor mRNA expression decreased while dopamine D2receptor mRNA expression increased in the lesioned side striatum of 6-OHDA-lesioned rats compared to control rats. Th ere were no signifi cant diff erences in MAO-B mRNA expression between control and 6-OHDA-lesioned rats. Th e pathological results demonstrated that there was a signifi cant decrease in the amount of hematoxylin and eosin stained neurons in lesioned side SNpc region of 6-OHDA-lesioned rats compared to the SNpc of control rats. These results are in accordance with related studies[19-26]. nAChRs desensitization following repeated nicotine stimulation could reverse signifi cantly the decrease of striatal dopamine and its metabolites and the increase in dopamine turnover in the lesioned side striatum of 6-OHDA-lesioned rats. Dopamine D1receptor mRNA expression increased significantly, whereas dopamineD2receptor mRNA expression remained unchanged in lesioned side striatum of nicotine-treated rats compared to 6-OHDA-lesiond rats when nAChRs were desensitized. Meanwhile, nicotine-treated rats exhibited a significant decrease in MAO-B mRNA expression in the lesioned side striatum compared to 6-OHDA-lesioned rats aft er nAChRs desensitization. However, there is no signifi cant change in the number of hematoxylin and eosin stained neurons and neurons morphology in the lesioned SNpc region between 6-OHDA-lesioned and nicotine-treated lesioned rats when nAChR is desensitized following repeated nicotine stimulation. Our data imply that desensitized nAChR could improve the decline in striatal dopaminergic function of 6-OHDA-lesioned rats through promoting dopamine release, upregulating dopamine D1receptor mRNA expression and downregulating MAO-B mRNA expression. These eff ects of desensitized nAChR are not induced by the alteration of the dopaminergic neurons damage in SNpc of hemiparkinsonian rats.

Fig. 3 Illustrative photomicrographs of coronal sections through the midbrain showing hematoxylin and eosin stained neurons in Control (A), 6-OHDA (B) and Nicotine+6-OHDA (C) groups on the left (lesion) side of SNpc. Average number of hematoxylin and eosin stained neurons in substantia nigra pars compacta (SNpc) in control, 6-OHDA-leisioned, and nicotine-treated lesioned groups(D). Nicotine was subcutaneously administered at 2.4 mg/kg/day for 14 days. Values are expressed as mean ± SEM, n=4,*P<0.05 vs controls. SNpc, substantia nigra compacta. Arrows, neurons in SNpc.
The finding that there is a negative correlation between cigarette smoking and the occurrence of PD has attracted a great many of studies to focus on the relationship between nicotine and PD. Previous studies mainly pay attention to the modulation of nicotine on the dopaminergic pathways and dopamine release[27-28]. In our previous study, we have observed the effects of different states of nAChR induced by diff erent doses of nicotine on rotational behavior in the unilateral 6-OHDA model of PD. We found that activated nAChRs did not exhibit protective role in behavioral impairment, while desensitized nAChRs signifi cantly improved the behavioral impairment[15]. Therefore, the advantage of our study on the relationship between nicotine and PD is that we discriminate diff erent states of nAChR using diff erent doses of nicotine and fi nd that desensitized nAChR can improve the decline in striatal dopaminergic function of 6-OHDA-lesioned rats through promoting DA release, upregulating dopamine D1receptor mRNA expression and downregulating MAO-B mRNA expression.
Th e pharmacologic action characteristics by which desensitized nAChRs improve the decline of striatal dopaminergic function in 6-OHDA-lesioned rats is that it could promote dopamine release, upregulate dopamine D1receptor mRNA expression and downregulate MAO-B mRNA expression, whereas has no significant influence on dopamine D2receptor mRNA expression. This action characteristic of desensitized nAChRs is different from that of current PD medications. Currently available therapeutics for PD includes administration of the dopamine precursor L-dopa and/or dopamine agonists[29-30]. Th ese drugs partially compensate for the decline in striatal dopamine that arises because of the loss of substantia nigra dopaminergic neurons. However, long-term dopamine replacement therapy is associated with the appearance of altered motor responses, including fl uctuations of the wearing-off type and dyskinesias that eventually become disabling[31]. Also, chronic levodopa therapy leads to psychiatric complications, and there is a loss of efficacy with time, probably because of disease progression, thereby limiting its long-term clinical application[32]. Most of current dopamine receptor agonists demonstrated poor selectivity for dopamine receptors thereby showing much side effect. Although dopamine selective agonists could selectively act on definite dopamine receptor subtype, they can induce receptor downregulation following long-term application, and then resulting in decrease of dopamine receptor function. Therefore, dopamine selective agonists are restricted in long-term clinical application due to above therapeutic contradiction. Desensitized nAChR selectively upregulate dopamine D1receptor mRNA expression, whereas do not influence dopamine D2receptor mRNA expression, consequently it has good selectivity on dopamine receptor subtype and refrain from the receptor downregulation issues which occurs during long-term application of dopamine selective agonists. Moreover, desensitized nAChRs could downregulate MAO-B mRNA expression, consequently avoiding the side eff ect resulting from poor selectivity of MAO inhibitors. In addition, studies from our laboratory have shown that desensitized nAChR could enhance dopamine receptor function, reduce rotational frequency of hemiparkinsonian rats. When brain nAChRs were in a subacute, acute or chronic desensitized state, induced by repeated administration of nicotine, activities of PKA and PKC were decreased. However, rats with hemiparkinsonism following unilateral 6-OHDA injection have a significant increase in PKA and PKC activities. Th erefore, based on the action characteristics by which desensitized nAChRs improve the decrease of dopaminergic function of 6-OHDA-lesioned rats, desensitized nAChR is a promising new therapeutic target that acts through pathways different from those of current PD medications and has distinguished advantages.
In summary, desensitized nAChRs could promote dopamine release, upregulate dopamine D1receptor
expression and downregulate MAO-B expression, thereby improve the decrease in striatal dopaminergic function of 6-OHDA-lesioned rats. These findings strongly suggest that desensitized nAChR is a promising new therapeutic pathway that acts through pathways distinct from those of current available therapeutics for PD and have distinguished predominance compared to current PD medications. We postulate that desensitized nAChR can be used as a new therapeutic target for the treatment of PD.
Acknowledgements
Th is work was supported by a grant from the National Natural Science Foundations of China (30371641).
1. Sauer H, Oertel WH. Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6-hydroxydopamine: a combined retrograde tracing and immunocytochemical study in the rat[J]. Neuroscience, 1994, 59(2): 401-415.
2. Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models[J]. Neuron, 2003, 39(6): 889-909.
3. Morens DM, Grandinetti A, Reed D, et al. Smoking-associated protection from Alzheimer’ s and Parkinson’s disease[J]. Lancet, 1994, 343(8893): 356-357.
4. Gorell JM, Rybicki BA, Johnson CC, et al. Smoking and Parkinson’s disease: a dose-response response relationship[J]. Neurology, 1999, 52(1): 115-119.
5. Damsma G, Day J, Fibiger HC. Lack of tolerance to nicotine-induced dopamine release in the nucleus accumbens[J]. Eur J Pharmacol, 1989, 168(3): 363-368.
6. Parain K, Marchand V, Dumery B, et al. Nicotine, but not cotinine partially protects dopaminergic neurons against MPTP-induced degeneration in mice[J]. Brain Res, 2001, 890(2): 347-350.
7. Karlin A. Emerging structure of the nicotinic acetylcholine receptors[J]. Nat Rev Neurosci, 2002, 3(2): 102-114.
8. Voitenko SV, Bobryshev AY, Skok VI. Intracellular regulation of neuronal nicotinic cholinoreceptors[J]. Neurosci Behav Physiol, 2000, 30(1): 19-25.
9. Nisell M, Nomikos GG, Hertel P, et al. Conditionindependent sensitization of locomotor stimulation and mesocortical dopamine release following chronic nicotine treatment in the rat[J]. Synapse, 1996, 22(4): 369-381.
10. Wang H, Sun XL. Desensitized nicotinic receptors in brain[J]. Brain Res Rev, 2005, 48(3): 420-437.
11. Liu Y, Wang Y, Sun XL, et al. Modulation of nicotine on functions and geneexpression of monoamine receptor in the brain[J]. Chin J Behav Med Sci, 2003, 12(6): 619-621.
12. Sun XL, Liu Y, Hu G, et al. Activities of cAMP-dependent protein kinase and protein kinase C are modulated by desensitized nicotinic receptors in the rat brain[J]. Neurosci Lett, 2004, 367(1): 19-22.
13. Cai G, Wang HY, Friedman E. Increased dopamine receptor signaling and dopamine receptor-G protein coupling in denervated striatum[J]. J Pharmacol Exp Th er, 2002, 302(3): 1105-1112.
14. Araki T, Tanji H, Kato H, et al. Alterations of second messenger systems in the rat brain after 6-hydroxydopamine lesions of the medial forebrain bundle[J]. Eur J Pharmacol Sci , 1999, 8(4): 261-267.
15. Han FR, Wang H. Effects of desensitized nicotinic receptors on rotational behavior in 6-hydroxydopamine model of Parkinson’s disease[J]. Neurosci Lett , 2007, 415(3): 200-204.
16. Paxinos G, Watson C. The rat brain in stereotaxic coordinates[M]. Academic Press, San Diego, CA, 1986.
17. Hudson JL, van Horne CG, Stromberg I, et al. Correlation of apomorphine- and amphetamine-induced turning with nigrostriatal dopamine content in unilateral 6-hydroxydopamine Lesioned rats[J]. Brain Res, 1993, 626(1-2): 167-174.
18. Rowell PP, Li M. Dose-response relationship for nicotine-induced up-regulation of rat brain nicotinic receptors[J]. J Neurochem, 1997, 68(5): 1982-1989.
19. Flores G, Liang JJ, Sierra A, et al. Expression of dopamine receptors in the subthalamic nucleus of the rat: characterization using reverse transcriptase-polymerase chain reaction and autoradiography[J]. Neuroscience, 1999, 91(2): 549-556.
20. Hurley MJ, Mash DC, Jenner P. Markers for dopaminergic neurotransmission in the cerebellum in normal individuals and patients with Parkinson’s disease examined by RTPCR[J]. Eur J Neurosci, 2003, 18(9): 2668-2672.
21. Joghataie MT, Roghani M, Negahdar F, et al. Protective effect of caffeine against neurodegeneration in a model of Parkinson’s disease in rat: behavioral and histochemical evidence[J]. Parkinsonism Relat Disord, 2004, 10(8): 465-468.
22. Ryoo HL, Pierrotti D, Joyce JN. Dopamine D3 receptor is decreased and D2 receptor is elevated in the striatum of Parkinson’s disease[J]. Mov Disord, 1998, 13(5): 788-797.
23. Robinet PM, Bardo MT. Dopamine D3 receptors are involved in amphetamine-induced contralateral rotation in 6-OHDA lesioned rats[J]. Pharmacol Biochem Behav, 2001, 70(1): 43-54.
24. Shachar DB, Kahana N, Kampel V, et al. Neuroprotection by a novel brain permeable iron chelator, VK-28, against 6-hydroxydopamine lesion in rats[J]. Neuropharmacology , 2004, 46(2): 254-263.
25. Xu Z, Cawthon D, McCastlain KA, et al. Selective alterations of gene expression in mice induced by MPTP[J]. Synapse, 2005, 55(1): 45-51.
26. Yuan H, Sarre S, Ebinger G, et al. Histological, behavioural and neurochemical evaluation of medial forebrain bundle and striatal 6-OHDA lesions as rat models of Parkinson’s disease[J]. J Neurosci Methods, 2005, 144(1): 35-45.
27. Benwell ME, Balfour DJ. Regional variation in the effects of nicotine on catecholamine overfl ow in rat brain[J]. Eur J Pharmacol, 1997, 325(1): 13–20.
28. Wonnacott S. Presynaptic nicotinic ACh receptors[J]. Trends Neurosci, 1997, 20(2): 92–98.
29. Olanow CW. Th e scientifi c basis for the current treatment of Parkinson’s disease[J]. Annu Rev Med, 2004, 55: 41–60.
30. Samii A, Nutt JG, Ransom BR. Parkinson’s disease[J]. Lancet, 2004, 363(9423): 1783-1793.
31. Chase TN, Engber TM, Mouradian MM. Contribution of dopaminergic and glutamatergic mechanisms to the pathogenesis of motor response complications in Parkinson’s disease[J]. Adv Neurol, 1996, 69: 497-501.
32. Goetz CG, Delong MR Richard MD. Neurosurgical horizons in PD[J]. Neurology, 1993, 43(1): 1-12.
doi 10.13459/j.cnki.cjap.2016.06.002
Hai WANG , MD, Professor, Cardiovascular Drug Research Center, Academy of Military Medical Sciences, Th adweik Academy of Medicine, Beijing 100850, China, Tel: 86-10-932651; Email: wh9588@sina.com.
2016-10-13; accepted 2016-11-11
杂志排行

中国应用生理学杂志的其它文章
- 5-HT1B受体亚型对小脑顶核介导的运动行为的影响*
- Association study between the angiotensin converting enzyme gene insertion/deletion polymorphism and Qinghai Han Chinese with congenital heart disease
- The infl uence of heterogeneity on the analysis of sleep-wake architecture in the single-prolonged stress rats
- Effect of creatine phosphate sodium on miRNA378, miRNA378* and calumenin mRNA in adriamycin-injured cardiomyocytes
- Changes of microcirculation in healthy volunteers and patients with septic shock in Xining
- 当归黄芪提取物对慢性腹膜功能衰竭大鼠腹膜功能、结构及TGF-β1表达的影响*