非典型溶血尿毒综合征
2016-06-01杨柳综述谢红浪审校
杨柳 综述 谢红浪 审校
·肾脏病临床·
非典型溶血尿毒综合征
杨柳 综述 谢红浪 审校
近来研究证实非典型溶血尿毒综合征(aHUS)与多种补体成分、活化因子及调节因子基因突变密切相关,基因筛查有助于病因诊断、预测预后及肾移植结局。补体H因子相关蛋白融合基因产物是aHUS发病机制的最新研究热点,抗补体治疗能够有效控制aHUS进展、改善预后,依库珠单抗与血浆置换、免疫抑制治疗、器官移植已被纳入aHUS治疗指南。对于尚未进入终末期肾病的患者进行单独肝移植是否普遍推广有待于进一步探索。本文旨在对aHUS诊治的最新研究进展作一综述。
非典型溶血尿毒综合征基因突变补体调节
溶血尿毒综合征(HUS)根据病因和临床表现分为典型HUS和非典型HUS(aHUS),后者指家族性或特发性补体替代途径调节异常所致HUS,与感染、药物、结缔组织病、器官移植、妊娠、代谢异常等因素有关的称之为继发性aHUS。儿童HUS中5%~10%为aHUS,而成人则以aHUS为主。约30%患者首次发病即进展至终末期肾病(ESRD),死亡率2%~10%。随着对aHUS患者补体基因突变的研究进展,原发性aHUS已用于专指补体调节异常所致的aHUS。本文主要就aHUS诊治的最新研究进展加以简述。
病因
正常人体内持续存在低水平C3活化裂解为C3a和C3b,C3a和C3b抑制C3裂解以避免补体旁路途径过度活化、攻击自身组织。aHUS患者存在补体替代途径失控,C3裂解产生C3b后结合B因子,C3Bb复合物即C3裂解酶能够促进更多C3b生成,新C3b片段结合C3转换酶后形成C5转换酶,作用于C5使其裂解为C5a和C5b,后者陆续结合C6、C7、C8和C9产生膜攻击复合体(MAC),造成内皮细胞损伤、血小板活化、炎症反应和血栓形成。该级联反应受多种蛋白调控,如I因子(FI)、H因子(FH)、血栓调节蛋白和膜辅蛋白(MCP)等(图1)。
近年来研究证实,aHUS与多种补体成分、活化因子及调节因子基因突变密切相关。1965年,Campbell等[1]最先报道一对同卵双生子发生溶血性贫血和氮质血症,随后陆续报道以儿童为先证者的家族性aHUS。1974年,人们首次发现aHUS患者血清补体C3水平下降而C4正常,且C3b、C3c、C3d等补体成分水平升高,提示存在补体替代途径活化[3]。1981年,Thompson等[4]报道一对aHUS患者兄弟均不产生补体替代途径的调节因子FH,而其近亲婚配的父母体内FH水平均仅为正常人的一半,表明此类患者存在遗传缺陷。1998年,研究发现aHUS与含有FH及其他补体调节因子基因的染色体1q32有关。后续基因学研究发现,50%~60% aHUS患者存在遗传性或获得性补体替代途径调节异常,其中20%携带编码补体替代途径主要调节因子FH的基因突变,另有10%患者抗FH自身抗体阳性[5-7](表1)。
目前已在aHUS患者发现补体H因子(CFH)及五种CFH相关蛋白(CFHR)基因异常的报道,CFH及CFHR1-5基因同源重复序列导致非等位基因同源重组、基因重排。4.5%aHUS患者存在CFH和CFHR杂合子基因重排,此类患者临床预后差、肾移植术后复发率高,Valoti等[8]发现5例携带CFH/ CFHR1基因的患者中有2例存在CFHR1/CFH融合基因,其蛋白产物含有4个短FHR1简易重复序列和20个FH简易重复序列,功能分析显示CFHR1/CFH融合基因产物能够竞争性拮抗FH并且诱导C5b-9沉积于内皮细胞表面,最终造成羊红细胞溶血。此结果表明CFHR1/CFH融合基因通过造成补体途径终末阶段过度活化导致内皮细胞破坏。aHUS患者中32%存在CFHR1纯合子缺失,25%检测到抗CFH抗体,而82%具有CFHR1纯合子缺失的患者为CFH抗体阳性,可见CFHR1纯合子缺失与抗FH抗体的形成密切相关[9]。

图1 补体替代途径[2]
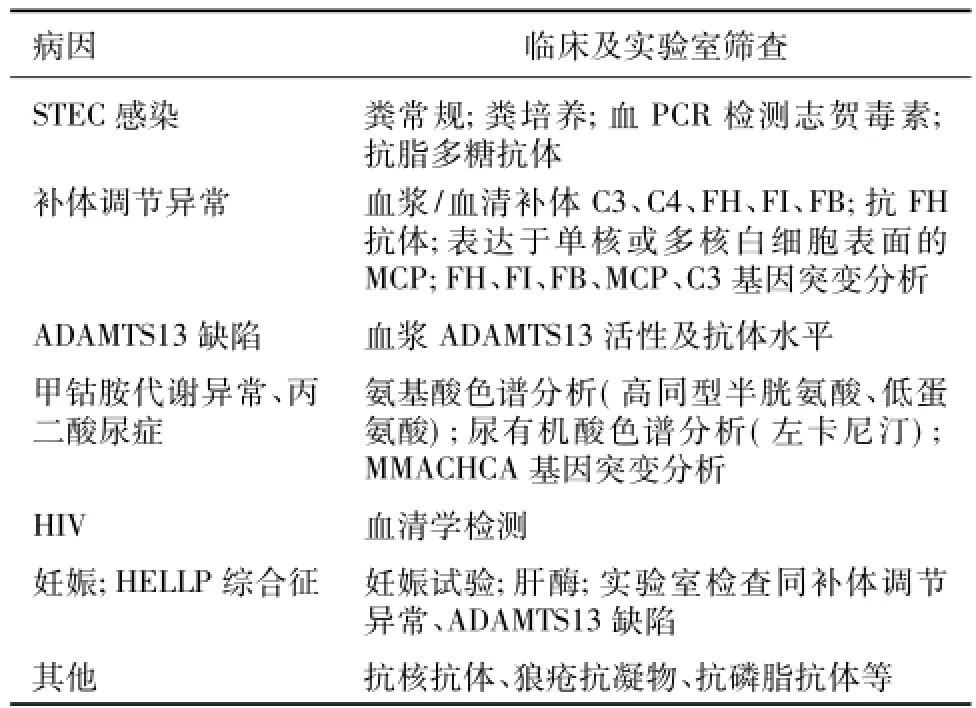
表1 非典型溶血尿毒综合征病因筛查[5]
诊断
由于aHUS和血栓性血小板减少性紫癜(TTP)临床表现相似,长期以来很难将两者明确区分。aHUS是补体成分或其调节因子基因异常或产生抗调节因子抗体造成补体替代途径过度活化,介导内皮水肿、内皮下间隙增宽并暴露胶原、vWF和纤维蛋白原,血小板活化形成血栓前状态,进一步造成广泛微血栓形成。而TTP发病主要基于vWF多聚体裂解蛋白酶ADAMTS13抑制物形成,导致ADAMTS13活性明显下降,vWF多聚体和血小板自发聚集引起形成微血栓。TTP患者多为抗ADAMTS13自身抗体作用于ADAMTS13抗原表位或半胱氨酸富集区和间隔区,导致ADAMTS13缺陷、vWF无法正常裂解。两者临床表现相互重叠,如血小板减少、外周血管微血栓形成和终末器官受损等,诊断均依赖临床表现和实验室检查,需排查感染、药物、器官移植、系统性红斑狼疮、抗磷脂抗体综合征、硬皮病、艾滋病、妊娠等诱因,通过粪培养、检测志贺毒素和抗脂多糖抗体排除典型HUS及测定ADAMTS13活性及抗体水平排除TTP等(表2)。
C3、C4、FH、FI、MCP、抗FH抗体以及CFH、补体I因子(CFI)、MCP、补体B因子(CFB)和C3突变的基因筛查是直接、有效地排查手段,应对所有外显子而非仅针对某些熟知位点进行筛查。Bresin等[6]通过基因筛查发现795例aHUS患者中44%存在基因突变(41%单基因突变,3%联合突变),具有CFH、C3或CFB突变的患者中8%~10%为联合突变,MCP或CFI突变者则有约25%存在联合突变,CFH或CFI突变者预后与联合突变无关。50%MCP联合突变患者起病3年内进展至ESRD,而MCP单基因突变者仅19%,MCP单基因突变者肾移植结局优于基因联合突变者。可见对于CFH、MCP单倍型和所有已知与aHUS相关基因进行分析有助于判断预后。
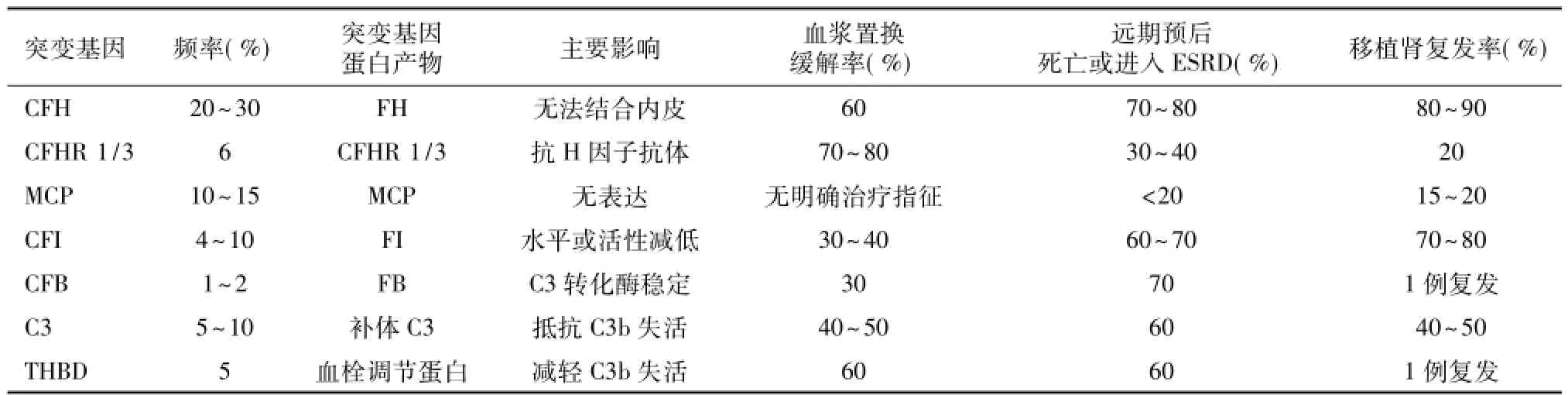
表2 非典型溶血尿毒综合征常见基因异常及临床结局[5]
治疗
血浆置换是治疗aHUS的一线疗法。通过清除抗FH自身抗体和过度活化的补体成分以及补充消耗的补体成分发挥作用。Igarashi等[10]于2014年发表的《溶血尿毒综合征诊治指南》建议确诊为aHUS后尽快进行血浆置换治疗,每次治疗成人需处理1~2倍血浆当量,儿童血浆量为50~100 ml/kg,推荐每日行血浆置换,至溶血控制后逐渐递减至每周3~5次。对血红蛋白低于60 g/L者应补充红细胞悬液,除非有严重出血倾向或大出血,否则不建议输注血小板。携带补体基因缺陷的个体多依赖血浆置换治疗,维持期需每周或每两周进行一次。除MCP基因突变外,其余补体基因突变均对血浆置换治疗反应良好[2,11]。
免疫抑制剂约6%~10%aHUS患者体内存在抗FH抗体,儿童发生率高于成人,以严重贫血、肝脏及中枢神经系统功能异常等肾外表现突出为特点,预后不佳。通常建议尽快给予血浆置换联合免疫抑制治疗,如激素冲击、环磷酰胺或利妥昔单抗等,直至抗FH抗体水平下降后切换为泼尼松及吗替麦考酚酯维持治疗。由于预后不良且存在有效的特异性治疗方法,起病即检测抗FH自身抗体具有重要意义,早期治疗者预后较好。
器官移植慢性肾脏病(CKD)5期患者可考虑肾移植,但由于aHUS复发率高,移植前需深入分析基因背景。研究表明FH、FI和C3基因突变者aHUS复发率分别高达75%~90%、45%~80%和40%~70%。Le等[12]对57例aHUS患者共计71次肾移植进行分析,发现CFH、CFI、MCP、C3以及CFB单基因突变占68%,移植肾5年存活率为51%,60%~70%患者因aHUS复发导致移植肾失功,其中70%发生于移植术后第一年内。存在CFH基因突变的个体复发率超过80%,而C3和CFB基因突变同样增加肾脏复发的风险,可见基因筛查是预测移植肾结局的重要方法[13]。对于MCP表达异常者,肾移植能够有效矫正补体缺陷、治疗aHUS,MCP突变者移植后复发率仅20%左右。亲属供肾移植后移植物失功几率高,因为供体可能存在相同基因突变而具aHUS复发风险,因此不推荐亲体肾移植。存在抗FH抗体者如果抗体水平持续偏低,则经肾移植术后免疫抑制治疗原发病复发几率较低。建议预防性血浆置换治疗以避免aHUS复发。
FH、FI、C3、FB均由肝脏合成,已进入ESRD且存在CFH或CFI突变的患者应考虑肝肾联合移植或肝移植,以有效修正补体调节蛋白基因缺陷,避免aHUS复发、移植肾失功。由于肝外来源的C3或CFB基因突变编码的蛋白可能导致aHUS肾脏复发,C3或CFB基因突变者是否适宜肝肾联合移植尚无定论。早年肝肾联合移植多因未采用血浆置换和缺血再灌注激活补体级联反应导致移植失败或移植肝失功。术前血浆置换及术中输注新鲜冰冻血浆明显改善了手术效果,目前已形成共识并在临床推广[14-15]。Haller等[16]报道了1例aHUS患者在肝肾损伤缓解后单独行肝移植,随访>2年,病情持续缓解。临床医师应慎重权衡对尚未进展至ESRD的aHUS患者行肝移植以保护肾脏的利弊,并对肝移植术后肾脏的情况进行长期观察。从短期效果来看,尽管单独肝移植风险较高,其对于提高生活质量和降低长期治疗成本仍具有重要意义。
补体调节依库珠单抗是重组人抗补体C5单克隆抗体,能够阻断C5裂解为C5a和C5b,于2009年被首次批准用于治疗aHUS[17]。研究表明依库珠单抗能够抑制补体终末阶段活化,减轻炎症和内皮损伤,改善凝血功能,促进肾脏修复[18]。无论是否涉及补体基因突变,依库珠单抗均能有效缓解病情,且早期使用能够更好地改善肾功能。临床试验证实依库珠单抗能够有效抑制补体异常介导的TMA,并显著改善aHUS患者的肾功能和生活质量,治疗过程中无严重感染,不良反应较少[19]。此外,依库珠单抗被成功用于治疗aHUS肾移植术后复发及明确基因突变者的术前预防性治疗[20-21]。指南建议一旦确诊aHUS应立即开始依库珠单抗治疗,且维持终身[22],但最近Wetzels等[23]报道3例aHUS患者病情稳定且肾功能改善持续4~6个月后,停用依库珠单抗至少4~6周,仅1例CFH外显子基因突变者出现复发,提示在部分患者在病情稳定后可尝试停药。FH异常引起的aHUS可输注重组纯化FH,相对血浆置换而言,纯化FH成本低,且无需建立中心静脉通路。目前已建立人血浆FH纯化技术,但尚未开展临床研究[24]。
小结:aHUS与补体系统异常活化密切相关,基因筛查对于病因诊断及预后判断具有重要意义。血浆置换、抗补体治疗能够缓解病情、改善预后,纯化补体FH有望成为新的治疗手段,但仍需大型临床试验进一步验证疗效及安全性。
1Campbell S,CarréIJ.Fatalhaemolyticuraemicsyndromeand idiopathic hyperlipaemia in monozygotic twins.Arch Dis Child,1965,40(214):654-658.
2Picard C,Burtey S,Bornet C,et al.Pathophysiology and treatment of typical and atypical hemolytic uremic syndrome.Pathol Biol(Paris),2015,63(3):136-143.
3Stühlinger W,Kourilsky O,Kanfer A,et al.Letter:Haemolytic-uraemic syndrome:evidence for intravascular C3 activation.Lancet,1974,2 (7883):788-789.
4Thompson RA,Winterborn MH.Hypocomplementaemia due to a genetic deficiency of beta 1H globulin.Clin Exp Immunol,1981,46 (1):110-119.
5Noris M,Remuzzi G.Atypical hemolytic-uremic syndrome.N Engl J Med,2009,361(17):1676-1687.
6Bresin E,Rurali E,Caprioli J,et al.Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype.J am Soc Nephrol,2013,24(3):475-486.
7Pérez-Caballero D,González-Rubio C,Gallardo ME,et al.Clustering of missense mutations in the C-terminal region of factor H in atypical hemolytic uremic syndrome.Am J Hum Genet,2001,68(2):478-484.8Valoti E,Alberti M,Tortajada A,et al.A novel atypical hemolytic uremic syndrome-associated hybrid CFHR1/CFH gene encoding a fusion proteinthatantagonizesfactorH-dependentcomplement regulation.J Am Soc Nephrol,2015,26(1):209-219.
9Hofer J,Janecke AR,Zimmerhackl LB,et al.Complement Factor HRelated Protein 1 Deficiency and Factor H Antibodies in Pediatric Patients with Atypical Hemolytic Uremic Syndrome.Clin J Am Soc Nephrol,2013,8(3):407-415.
10 Igarashi T,Ito S,Sako M,et al.Guidelines for the management and investigation of hemolytic uremic syndrome.Clin Exp Nephrol,2014,18(4):525-557.
11 Taylor CM,Machin S,Wigmore SJ,et al.Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom.Br J Haematol,2010,148(1):37-47.
12 Le Quintrec M,Zuber J,Moulin B,et al.Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome.Am J Transplant,2013,13(3):663-675.
13 Loirat C,Fremeaux-Bacchi V.Hemolytic uremic syndrome recurrence after renal transplantation.Pediatr Transplant,2008,12(6):619-629.
14 Saland JM,Emre SH,Shneider BL,et al.Favorable long-term outcome after liver-kidney transplant for recurrent hemolytic uremic syndrome associated with a factor H mutation.Am J Transplant,2006,6(8): 1948-1952.
15 Saland JM,Ruggenenti P,Remuzzi G;Consensus Study Group.Liverkidney trans-plantation to cure atypical hemolytic uremic syndrome.J Am Soc Nephrol,2009,20(5):940-949.
16 Haller W,Milford DV,Goodship TH,et al.Successful isolated liver transplantation in a child with atypical hemolytic uremic syndrome and a mutation in complement factor H.Am J Transplant,2010,10(9): 2142-2147.
17 Rother RP,Rollins SA,Mojcik CF,et al.Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria.Nat Biotechnol,2007,25(11):1256-1264.18 Cofiell R,Kukreja A,Bedard K,et al.Eculizumab reduces complement activation,inflammation,endothelial damage,thrombosis,and renal injury markers in aHUS.Blood,2015,125(21):3253-3262.
19 Legendre CM,Licht C,Muus P,et al.Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome.N Engl J Med,2013,68(23):2169-2181.
20 Zuber J,Le Quintrec M,Sberro-Soussan R,et al.New insights into postrenal transplant hemolytic uremic syndrome.Nat Rev Nephrol,2011,7(1):23-35.
21 Nester C,Stewart Z,Myers D,et al.Pre-emptive eculizumab and plasmapheresis for renal transplant in atypical hemolytic uremic syndrome.Clin J Am Soc Nephrol,2011,6(6):1488-1494.
22 Wong EK,Goodship TH,Kavanagh D.Complement therapy in atypical haemolytic uraemic syndrome(aHUS).Mol Immunol,2013,56(3): 199-212.
23 WetzelsJF,vandeKarNC.Discontinuationofeculizumab maintenance treatment for atypical hemolytic uremic syndrome.Am J Kidney Dis,2015,65(2):342.
24 Brandst tter H,Schulz P,Polunic I,et al.Purification and biochemical characterization of functional complement factor H from human plasma fractions.Vox Sang,2012,103(3):201-212.
Recent advances in atypical hemolytic uremic syndrome
YANG Liu,XIE Honglang
National Clinical Research Center of Kidney Diseases,Jinling Hospital,Nanjing University School of Medicine,Nanjing 210016,China
Recently,studies confirmed with the relationship between atypical Hemolytic uremic syndrome (aHUS)and gene mutations of many complement factors,activators and regulators.Gene screening is helpful for diagnosis and prognosis and predicts the outcome of renal transplantation.New focus on pathogenesis of aHUS is fusion protein product of CFH related protein 1(CFHR1)/CFH rearrangements.Clinical trials show that proper complement regulation treatment could control aHUS development and improve prognosis.Eculizumab,plasma exchange,immunosuppressive therapy and organ transplantation are included in aHUS therapeutic guideline.Liver transplantation for patients before end stage renal disease needs further exploration.This review summarizes progression of diagnosis and treatment of aHUS.
atypicalhemolytic uremic syndromegene mutationcomplement regulation
2015-05-25
(本文编辑凡心)
10.3969/cndt.j.issn.1006-298X.2016.01.016
南京军区南京总医院肾脏科国家肾脏疾病临床医学研究中心全军肾脏病研究所(南京,210016)
