高血压、蛋白尿、肾脏病家族史
2016-06-01范文静葛永纯程震刘志红
范文静 葛永纯 程震 刘志红
·临床集锦·
高血压、蛋白尿、肾脏病家族史
范文静 葛永纯 程震 刘志红
40岁男性患者,以眼睑水肿、高血压、蛋白尿起病,肾功能正常,血清载脂蛋白A-I水平下降。患者父亲因“肾脏病”去世。肾活检肾组织刚果红染色阳性,皮肤脂肪组织及肠黏膜刚果红染色阴性。电镜下肾小球系膜区及内皮下大量异常物质沉积,肾小管间质及血管未受累。肾组织载脂蛋白A-Ⅰ(ApoA-Ⅰ)染色阳性,肾小球微分离质谱分析见高浓度ApoA-Ⅰ条带,基因检测结果显示ApoA-Ⅰ基因c.220A>G突变。该患者最终诊断为遗传性系统性载脂蛋白A-Ⅰ淀粉样变性。
遗传性系统性淀粉样变载脂蛋白A-I肾活检蛋白尿
病史摘要:
现病史40岁男性患者,因“尿检异常1年余”,于2014-12-30入院。
患者于2013年7月饮酒后出现双踝及眼睑水肿,当地医院查尿蛋白++,红细胞2.8个/HP,肾活检示“肾组织刚果红染色阳性,电镜下见肾小球系膜区、毛细血管袢基膜外可见大量淀粉样纤维沉积”,诊断为“淀粉样变性肾病”。2013年8月患者就诊于北京某医院行重复肾活检光镜示“55个肾小球,肾小球系膜区大量刚果红和偏振光阳性的特殊蛋白沉积,基底膜轻度增厚”;免疫组化染色“A蛋白-,TTR-,Fibrinogen+,Lys±”,肾小球微分离行蛋白质质谱分析见高浓度遗传性载脂蛋白A-Ⅰ(ApoA-Ⅰ)条带,最终诊断为“肾淀粉样变性,遗传性纤维蛋白(Fibrinogen)淀粉样变性可能性大”。患者2014年12月入住我科,病程中无消瘦、低血压及夜间不能平卧,体重无明显变化,尿量正常。
既往史发现高血压1年余,最高150/90 mmHg,目前服福辛普利、硝苯地平控释片降压,血压控制于120~130/70~80 mHg;其余无特殊。
家族史父亲60岁去世,有“肾病综合征”病史3年。母健在,1兄1妹及1女查尿常规均正常。其他家族成员否认肾脏病史。
体格检查发育正常,体质量指数(BMI)20.0 kg/m2,血压142/80 mmHg,右眼鼻侧结膜可见轻度凸出异常物质沉积,无视物模糊。心肺腹部查体未及明显异常,双下肢无水肿。
实验室检查
尿液尿蛋白定量2.11~3.52 g/24h,尿沉渣红细胞计数2万/ml,尿白细胞0~1/HP;α2-MG 2 mg/L,C3 3 mg/L,RBP 0.5 mg/L,NAG酶25.5 U/(g·Cr),尿渗量482 mOsm/(kg·H2O)。
血液血红蛋白114 g/L,白细胞计数7.5×109/L,血小板193×109/L,网织红细胞百分数1.09%。白蛋白32.3 g/L,球蛋白16.7 g/L,血清肌酐80 μmol/L,尿素氮4.9 mmol/L,尿酸430 μmol/L,胱抑素C 1.17 mg/L,总胆固醇6.81 mmol/L,三酰甘油1.97 mmol/L,HDL 0.42 mmol/L,LDL 4.79 mmol/L,载脂蛋白AI 0.58 g/L (参考范围1.0~5.0 g/L),载脂蛋白B 1.25 g/L,载脂蛋白E 4.02 mg/dl,谷丙转氨酶/谷草转氨酶21/21 U/L,前白蛋白、乳酸脱氢酶、胆红素均正常。估算的肾小球滤过率(eGFR)106.46 ml/(min·1.73m2)。
心功能指标N-端前脑钠肽、脑利钠肽前体、肌钙蛋白T、肌钙蛋白I、肌酸激酶MB同工酶、肌酸激酶MB同工酶质量均正常。
肿瘤标志物甲胎蛋白2.02 μg/L、β2-MG 2.28 mg/L。
甲状腺功能TSH 6.35 mIU/L,余正常,甲状腺抗体阴性。
免疫学ANA、抗ds-DNA抗体、抗磷脂酶A2受体抗体阴性,血游离轻链:κ 53.94 mg/L,λ 48.98 mg/L,κ/λ 1.10,免疫固定电泳未见单克隆条带,IgG 4.330 g/L、IgA 1.380 g/L、IgM 0.884 g/L、IgE 29.7 IU/ml、RF<20.0、ASO 28.7 IU/ml,补体正常,外周血淋巴细胞亚群CD4 1 224个/μl,CD8 850个/μl。
骨髓细胞学检查大致正常骨髓象。
辅助检查
双肾B超左肾112 mm×48 mm×56 mm,右肾110 mm×35 mm×55 mm,皮质厚度不清,皮质回声增强,皮髓界限清楚,集合系统正常。左肾上极见一大小约17 mm×17 mm的类圆形无回声区,界清。双肾轮廓规则,包膜连续完整,双肾内未见肾盂肾盏扩张。
胸片、甲状腺超声、颈部及双下肢血管超声未见异常。
消化系统超声脾大4.8 cm×13.7 cm。
心电图窦性心动过缓、左心室高电压、T波改变。
心脏超声房间隔中部可见极少量左向右过隔血流,考虑卵圆孔未闭。室间隔厚度9 mm,左心室内径49 mm,LVEF:54%。
眼底检查视盘色清,色淡红,A/V 1∶2,后极部网膜平伏,未见明显出血及渗出。右眼鼻侧巩缘处结膜异常增厚隆起(图1),其下见黄白色物质沉积,未累及角膜。
咽喉视诊正常。
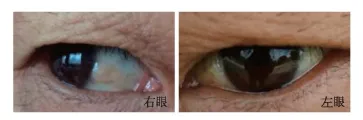
图1 患者右眼鼻侧巩缘处黄白色物质沉积
肾活检病理
光镜正切肾小球体积增大,肾小球固有细胞明显减少,系膜区增宽,系膜区及内皮下大量PAS、HE均质淡染物质沉积,袢腔几近堵塞,囊壁节段增厚(图2A)。PASM-Masson:上述沉积物不嗜银,嗜亮绿(图2B)。肾小管间质慢性病变轻度,灶性小管萎缩、基膜增厚,管腔内见蛋白管型,间质灶性单个核细胞浸润,小动脉偶见透明变性。
刚果红染色肾小球内大量橘红色物质沉积(图2C),偏振光下呈苹果绿双折光,肾间质及肾血管刚果红染色阴性。
免疫荧光IgG+,κ轻链+、λ轻链+,呈颗粒状沉积于肾小球系膜区和血管袢,IgA、IgM、C3、C1q阴性。荧光染色ApoA-Ⅰ阳性(图2D),呈团块状弥漫分布于系膜区。ApoB,ApoE阴性。Lysozyme、APP、transthyretin、β2-MG、LECT2染色阴性。
电镜肾小球系膜区增宽,系膜区及系膜旁区低倍镜下见大量无定形的中等偏低电子密度的物质,高倍镜下见排列紊乱无分枝的细纤维丝,直径8~14 nm。肾小球系膜区及毛细血管袢基膜内皮下、上皮侧见散在分布、中~高电子密度的致密物沉积(2E),高倍镜下无特殊结构(图2F)。肾小球毛细血管袢基膜厚270~580 nm。足细胞足突融合50%~60%。
皮肤脂肪、肠黏膜病理刚果红染色阴性。
基因分析患者基因检测结果显示ApoA-Ⅰ基因编码区第220位碱基由A突变为G,引起该基因编码的氨基酸序列第74位置上,氨基酸W突变为氨基酸R。
诊断分析中年男性,病史特点为:(1)高血压、蛋白尿起病,伴间断水肿;(2)肾损害表现中等量蛋白尿,无镜下血尿,轻度低白蛋白血症,肾功能正常,肾外见球结膜赘生物形成,无低血压等肾外表现;(3)父亲因肾病综合征去世;(4)外院曾2次行肾活检,肾组织刚果红染色阳性,电镜见淀粉样纤维沉积;(5)我院检查血游离轻链比值、绝对值及免疫固定电泳正常,重复肾活检示肾小球刚果红染色阳性,肾间质及肾血管刚果红染色阴性,心脏、皮肤脂肪及肠黏膜评价未见受累。综上所述,患者肾淀粉样变性诊断明确,可排除AL型、AA型淀粉样变性,诊断及鉴别诊断围绕遗传性淀粉样变性可能。
遗传性淀粉样变性患者通常血游离轻链绝对值及比值正常,M蛋白检出率低,无显著心脏受累及低血压表现。不同的淀粉样物质前体蛋白引起的遗传性淀粉样变性的临床及病理表现各不相同。溶菌酶型淀粉样变性常累及消化道,可发生肝破裂、出血及胃肠道出血,肾损害表现为高血压、肾病综合征,淀粉样物质可沉积于肾小球、肾小管间质及血管;甲状腺素结合蛋白淀粉样变性以显著的外周神经病变及心肌病变为特点,肾脏淀粉样变性较少见;纤维蛋白原Aα淀粉样变性患者可表现为高血压、蛋白尿,肾脏病理可见淀粉样物质主要沉积于肾小球,极少累及血管和肾小球间质;ApoA-Ⅰ淀粉样变性的表现多样,肾脏及肝脏的受累较为多见,通常不累及心脏。
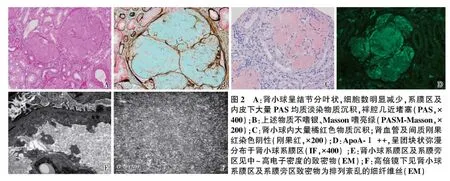
图2A:肾小球呈结节分叶状,细胞数明显减少,系膜区及内皮下大量PAS均质淡染物质沉积,袢腔几近堵塞(PAS,× 400);B:上述物质不嗜银、Masson嗜亮绿(PASM-Masson,× 200);C:肾小球内大量橘红色物质沉积;肾血管及间质刚果红染色阴性(刚果红,×200);D:ApoA-Ⅰ++,呈团块状弥漫分布于肾小球系膜区(IF,×400);E:肾小球系膜区及系膜旁区见中~高电子密度的致密物(EM);F:高倍镜下见肾小球系膜区及系膜旁区致密物为排列紊乱的细纤维丝(EM)
本例患者肾脏病理特点以肾小球受累为主,小管间质及血管无淀粉样物质沉积,因此纤维蛋白原Aα淀粉样变性及ApoA-Ⅰ淀粉样变性均有可能。因患者于外院行肾小球微分离质谱分析见高浓度ApoA-Ⅰ条带,所以我们考虑ApoA-Ⅰ淀粉样变性可能性大。为验证此推测,对患者肾组织切片行ApoA-Ⅰ、ApoB、ApoE、Lysozyme、transthyretin等特殊淀粉样物质染色,结果仅见ApoA-Ⅰ染色阳性。对患者及亲属基因检测分析结果见患者ApoA-Ⅰ基因突变,最终得以明确诊断。
最后诊断(1)ApoA-Ⅰ淀粉样变性(累及肾脏、肝脏、球结膜);(2)肾淀粉样变性CKD1期。
治疗及随访诊断明确后予骨化三醇、非诺贝特、血管紧张素转换酶抑制剂(ACEI)及虫草制剂等对症治疗,嘱其低脂、优质蛋白饮食。5月后复查转氨酶正常,血清肌酐70.7 μmol/L,血清白蛋白无明显变化,球蛋白升高至25.2 g/L,尿蛋白++。
讨论
该例中年男性患者,病程1年余,多器官受累(肾脏、肝脏、球结膜),肾活检结果结合患者家族史、基因检测结果,最终患者明确诊断为ApoA-Ⅰ淀粉样变性。
ApoA-Ⅰ淀粉样变性最初由1969年Van Allen等[1]报道,目前国际上已有上百例报道。目前认为是一类常染色体显性遗传疾病,发病年龄18~65岁,多于中年时期出现临床症状。不同基因突变类型累及脏器不同,以肾脏、肝脏最为常见,其他还包括脾脏、外周神经、胃肠道、皮肤、喉部、生殖腺、视网膜等。ApoA-Ⅰ由肝脏和小肠合成,分子量28 300D,编码ApoA-Ⅰ的基因定位于11号常染色体q23-q24,成熟的ApoA-Ⅰ蛋白由ApoA-Ⅰ基因的外显子3、4编码的243个氨基酸组成[2]。ApoA-Ⅰ基因突变热点区域为50至93位点,170至178位点。目前已报道的19种ApoA-Ⅰ基因突变(表1)[3-10],多数为核苷酸替代突变,以及3个移码突变(均以肾脏受累为主),2个缺失突变,1个缺失/插入突变。Gly26Arg突变为最常见类型,即26位精氨酸替代甘氨酸,表现为突出的肾脏及外周神经受累。50-93位点的突变常引起肾脏和肝脏受累,而170-178位点突的变更倾向于出现心脏、喉及皮肤的受累[10]。

表1 现已报道的ApoA-Ⅰ基因突变
从表1可见,以肾脏受累为主要表现的ApoA-Ⅰ淀粉样变性患者一般无心肌淀粉样物质沉积,但ApoA1变异体使HDL功能受限,引起血胆固醇升高、动脉粥样硬化所致心血管事件却是ApoA-Ⅰ淀粉样变性患者死亡的主要原因之一[11]。该淀粉样物质在肝脏沉积表现为肝大、肝功能异常。外周神经受累表现为相应的神经受累。Glu34Lys突变的患者还可见结膜异常物质沉积[12]。
既往认为ApoA-Ⅰ淀粉样变性以单纯累及肾小球为特点,肾小管间质及血管不受累。但基因突变类型不同,病理改变也不尽相同。近期Gregorini等[8]报道了对50个ApoA-Ⅰ基因Leu75Pro突变所致淀粉样变性患者及其家族研究,结果显示135个突变携带者至少有肾脏、肝脏、睾丸中一个器官的受累。肾脏是最常见的首发受累器官,起病初即可有血清肌酐高,尿液分析通常阴性,或仅为少量小管性蛋白尿,21例患者肾活检见管周及间质淀粉样物质沉积,ApoA-Ⅰ染色阳性。肾小球的损伤很大程度上继发于小管硬化,刚果红染色阴性。提示ApoA-ⅠLeu75Pro基因突变的淀粉样变性患者以肾小管淀粉样物质沉积为肾脏病理表现。
遗传性淀粉样变性是基因异常所致的疾病,尚无明确有效治疗方案,遗传性甲状腺素转运蛋白淀粉样变性患者肝脏受累者行肝移植可获得较好的预后,这可能由于甲状腺素转运蛋白是由肝脏产生[13]。ApoA-Ⅰ由肝脏和小肠共同合成,故肝肾移植治疗对ApoA-Ⅰ淀粉样变性患者仅可获得一定程度的缓解,而非完全缓解。本例患者肾小球大量ApoA-Ⅰ蛋白沉积,血ApoA、LDL低水平,患者2014年12月重复肾活检病理与2013年外院肾活检结果相比较,肾小球血管袢腔几近完全堵塞,刚果红阳性物质所占肾小球面积明显增加,肾脏病理、病情发展较迅速。我们最终予患者骨化三醇口服治疗,以期减少异常ApoA-Ⅰ的产生,其原理基于G revik等[14]的实验观察发现,使用大剂量睾酮后的健康志愿者中,其血脂谱ApoA-Ⅰ及HDL脂蛋白水平明显下降,其原因可能为长期使用类固醇激素的人体内25羟-维生素D水平变化可引起血浆中ApoA-Ⅰ水平下降。同时予非诺贝特、ACEI降压及虫草制剂治疗。5月后随访,患者血清肌酐呈轻度下降,尿蛋白及血白蛋白未见进一步恶化。但由于治疗时程短,其疗效、长期预后以及可能的最佳治疗方案,尚待进一步观察。
随着对淀粉样变性认识的加深,肾活检病理、免疫病理特殊染色技术的不断进展,淀粉样变性的诊断率越来越高,分型日渐精确。但特殊荧光染色尚存在不足,表现在技术手段、染色方法以及主观判断的误差,均可造成误诊、漏诊,特殊类型淀粉样变性被漏诊、误诊,均可带来后续治疗方案的失败。免疫组织化学技术是肾活检病理诊断的重要手段,在免疫病理观察过程中,还应区分免疫球蛋白和补体的非特异性和特异性沉积。节段硬化及球性硬化的肾小球中,常见血浆内渗或血浆蛋白漏至硬化区,以IgM和C3较为常见。本例患者曾在外院诊断为“遗传性纤维蛋白(Fibrinogen)淀粉样变性”,而我院复查Fibrinogen染色则为阴性,且临床表现、基因检测结果均不符合该疾病。
本例患者的诊断过程中,蛋白组学分析和基因检测手段发挥了较大的作用。蛋白组学技术通过系统性地分析和鉴定血液、尿液中蛋白质分子并研究其生物功能,为临床疾病的诊断提供了重要线索和方向;基因检测手段通过连锁分析对染色体片段进行疾病区点定位,使基因缺陷疾病得到准确诊断,是一种无创、高效的疾病诊断方法,并为临床用药、优生优育提供重要信息。尤其在肾脏结构不佳、穿刺风险大以及不能接受肾活检的患者中,基因检测可为明确病因、准确诊断、指导治疗、提示预后,以及判断家族疾病基因携带情况提供较为可靠的答案。
小结:本文报道了国内首例确诊为ApoA-Ⅰ淀粉样变性的患者,并结合国外相关文献,分析该疾病的临床、病理特点,及诊疗过程,以期加深对该疾病的认识,提高诊断率。
1Van Allen MW,Frohlich JA,Davis JR.Inherited predisposition to generalized amyloidosis.Clinical and pathological study of a family with neuropathy,nephropathy,and peptic ulcer.Neurology,1969,19 (1):10-25.
2Brewer HB Jr,Fairwell T,LaRue A,et al.The amino acid sequence of human ApoA-Ⅰ,anapolipoproteinisolatedfromhighdensity lipoproteins.Biochem Biophys Res Commun,1978,80(3):623-630.
3Booth DR,Tan SY,Booth SE,et al.A new apolipoprotein Al variant,Trp50Arg,causes hereditary amyloidosis.QJM,1995,88(10): 695-702.
4Soutar AK,Hawkins PN,Vigushin DM,et al.Apolipoprotein AI mutation Arg-60 causes autosomal dominant amyloidosis.Proc Natl Acad Sci USA,1992,89(16):7389-7393.
5Murphy CL,Wang S,Weaver K,et al.Renal apolipoprotein A-I amyloidosis associated with a novel mutant Leu64Pro.Am J Kidney Dis,2004,44(6):1103-1109.
6Caballería J,Bruguera M,Solé M,et al.Hepatic familial amyloidosis caused by a new mutation in the apolipoprotein AI gene:clinical and pathological features.Am J Gastroenterol,2001,96(6):1872-1876.
7Eriksson M,Sch nland S,Yumlu S,et al.Hereditary apolipoprotein AI-associatedamyloidosisinsurgicalpathologyspecimens: identification of three novel mutations in the ApoA1 gene.J Mol Diagn,2009,11(3):257-262.
8Gregorini G,Izzi C,Ravani P,et al.Tubulointerstitial nephritis is a dominant feature of hereditary apolipoprotein A-I amyloidosis.Kidney Int,2015,87(6):1223-1229.
9Hazenberg AJ,Dikkers FG,Hawkins PN,et al.Laryngeal presentation of systemic apolipoprotein A-I-derived amyloidosis.Laryngoscope,2009,119(3):608-615.
10 Rowczenio D,Dogan A,Theis JD,et al.Amyloidogenicity and clinical phenotype associated with five novel mutations in apolipoprotein A-I.Am J Pathol,2011,179(4):1978-1987.
11 Hamidi Asl L,Liepnieks JJ,Hamidi Asl K,et al.Hereditary amyloid cardiomyopathy caused by a variant apolipoprotein A1.Am J Pathol,1999,154(1):221-227.
12 Andeen NK,Lam DY,de Boer IH,et al.Renal ApoA-1 amyloidosis with glu34lys mutation and intra-amyloid lipid accumulation.J Am Soc Nephrol,2014,25(12):2703-2705.
13 Joy T,Wang J,Hahn A,et al.ApoAⅠrelated amyloidosis:a case report and literature review.Clin Biochem,2003,36(8):641-645.
14 G revik N,Rane A,Bj rkhem-Bergman L,et al.Effects of different doses of testosterone on gonadotropins,25-hydroxyvitamin D3,and blood lipids in healthy men.Subst Abuse Rehabil,2014,5:121-127.
Hypertension,proteinuria and a family history of kidney disease
FAN Wenjing,GE Yongchun,CHENG Zhen,LIU Zhihong
National Clinical Reserch Center of Kidney Diseases,Jinling Hospital,Nanjing University School of Medicine,Nanjing 210016,China
A 40-years-old male patients who had a family history of kidney disease was admitted for hypertension,proteinuria and recurrent edema with normal renal function.Congo red staining was positive in his renal tissue but negative in skin,subcutaneous fat tissue and rectal mucosa.Those amyloid substances were deposited in mesangium and subendothelial area but absent in tubulo-interstitial area and vessels.Further exploration for the precursor protein showed positive staining of ApoA-Ⅰand negative results for light chains,ApoB,ApoE,Lysozyme,transthyretin,and β2-microglobulin.Sequencing of the apolipoprotein A-Ⅰgene revealed a mutation of c.220A>G.Finally he was diagnosed hereditary apolipoprotein A1 amyloidosis.
hereditary amyloidosisapolipoprotein A-Ⅰrenal biopsyproteinuria
2015-08-27
(本文编辑律舟莫非)
10.3969/cndt.j.issn.1006-298X.2016.01.021
南京军区南总医院肾脏科国家肾脏疾病临床医学研究中心全军肾脏病研究所(南京,210016)
