遗传性间质性肾炎致病基因研究进展
2016-06-01乔盼盼综述王朝晖审校
乔盼盼 综述 王朝晖 审校
·医学继续教育·
遗传性间质性肾炎致病基因研究进展
乔盼盼 综述 王朝晖 审校
近来遗传性间质性肾炎致病基因的研究取得一些进展,已发现常染色体隐性遗传的肾单位肾痨(NPHP)致病基因20种。另外,改善全球肾脏病预后组织(KDIGO)提出了常染色体显性遗传性肾小管间质性肾病(ADTKD)的新定义及其诊断标准[1],现已发现4种ADTKD的致病基因(MUC1,UMOD,REN,HNF1B)。通过对上述致病基因的研究,探讨其可能导致的病理改变及临床症状,对遗传性间质性肾炎的预防、基因诊断、治疗、预后具有重要意义。
肾单位肾痨常染色体显性遗传性肾小管间质性肾病肾髓质囊性病致病基因
遗传性间质性肾炎是一组主要涉及肾小管间质损伤的疾病,与致病基因突变相关、具有家族聚集和遗传倾向[2],主要有常染色体隐性遗传和常染色体显性遗传两种遗传方式,前者主要指肾单位肾痨(NPHP),后者指常染色体显性遗传性肾小管间质性肾病(ADTKD)。本文主要对NPHP及ADTKD致病基因的相关研究进展进行综述。
NPHP
1951年Fanconi等[3]首次提出NPHP,该病是导致儿童和青少年终末期肾病(ESRD)最常见的遗传性疾病。临床常见烦渴、多尿、贫血、生长迟缓等,组织病理学改变无特异性,主要为肾小管间质性改变:肾小管萎缩、肾小管基膜增厚或变薄、间质纤维化及炎细胞浸润[4]。NPHP早期症状轻微,可进展为ESRD,部分患者出现严重贫血、生长发育迟缓及高血压等。根据其发展为ESRD的平均时间可分为:少年型即NPHP1型(7~25岁)、婴儿型即NPHP2型(1~3岁)、青年型即NPHP3型(11~28岁),以少年型最为常见。NPHP临床表现无特异性,与部分肾脏疾病(如原发性高草酸尿症)的临床表现相似[5]。肾脏超声可见肾脏大小正常或萎缩,回声增强,皮髓质分界不清,晚期可见肾脏皮髓质交界区囊肿,但是婴儿型(NPHP2型)表现为肾脏稍增大,组织病理学同时出现NPHP的特征性改变(间质纤维化和肾小管萎缩)和多囊肾病的病理改变(肾脏体积增大及广泛性囊肿形成),因此NPHP2型易与多囊肾相混淆。约15%的NPHP合并有肾外症状,主要包括视网膜变性(Senior-Loken综合征)、小脑蚓部发育不全(Joubert综合征)、肝纤维化、内脏转位以及锥形骨骺等。
NPHP的诊断依赖肾脏活检或基因检测。自1997年发现致病基因NPHP1后,现已发现20种致病基因(NPHP1-18,NPHP1L,NPHP2L)及与之相关的综合征(表1)。

表120 种NPHP致病基因一览[4-15]
ADTKD
ADTKD是一组单基因遗传性肾小管间质性肾病,其致病基因包括MUC1、UMOD、REN和HNF1B,分别对应ADTKD的子分类:ADTKD-MUC1; ADTKD-UMOD;ADTKD-REN和ADTKD-HNF1B。
ADTKD-MUC1以往称肾髓质囊性病1型(MCKD1),MCKD为常染色体显性遗传的肾小管间质性疾病,与NPHP具有相似的临床表现及病理组织特点,因此常将MCKD与NPHP合称肾单位肾痨-肾髓质囊性病(NPHP-MCKD),但MCKD主要为成人起病,按照致病基因可分为MCKD1型和MCKD2型,两者致病基因分别为MUC1和UMOD,但是无论是MCKD1型还是MCKD2型,其临床影像显示的肾小管囊样扩张甚至较大的囊肿及肾髓质改变均不是特异性诊断指标,“肾髓质囊性病”这一名称易引起临床医师误解,所以KDIGO建议更改此类疾病名称为ADTKD-MUC1和ADTKD-UMOD。MUC1定位于染色体1q21,编码膜锚定黏蛋白mucin1,此蛋白在多种组织细胞(皮肤、乳腺、肺脏、胃肠道、唾液腺及肾脏远端小管细胞)均有表达,可能形成细胞表面保护层并参与细胞信号传导过程,但是MUC1突变引起的ADTKD-MUC1并未发现有除肾脏外其他器官病变[16]。研究发现MUC1基因的VNTR域中一个胞嘧啶重复复制可引起移码突变,编码突变蛋白MUC1-fs,此种突变蛋白含有大量碱性氨基酸,可导致折叠障碍,所以容易在肾小管上皮细胞内沉积[17],引起肾小管细胞凋亡。
ADTKD-UMOD以往称尿调节素相关性肾病(UAKD)、肾髓质囊性病2型(MCKD2)及家族性少年型高尿酸血症肾病1型(FJHN1),FJHN为常染色体显性遗传的肾小管间质性疾病,1960年由Duncan和Dixon等[18]首次描述,现已发现其致病基因有:UMOD、REN,按致病基因分别命名为FJHN1型和FJHN2型。MCKD2型与FJHN1型在遗传方式、临床表现、病理组织改变等方面有许多共同之处。2000年,Dahan等[19]提出MCKD2型和FJHN1型属于同一种疾病,现KDIGO建议更改此类疾病名称为ADTKD-UMOD,致病基因为UMOD,定位于染色体16p12.3,在肾小管初级纤毛中表达,编码尿调节素(uromodulin)又称Tamm-Horsfall蛋白(THP),是正常人尿液中含量最多的蛋白质,由髓绊升支粗段肾小管上皮细胞特异生成,除一些信号肽及磷脂酰肌醇共有序列之外,其590个氨基酸残基中包含480个半胱氨酸残基,半胱氨酸残基之间依靠二硫键连接保持蛋白质稳定性。UMOD共有11个外显子,其中外显子2-11编码尿调节素,2009年Williams等证实UMOD突变存在于外显子3、4、5和外显子7[20]。外显子3和外显子4编码类似表皮生长因子的结构域和一个半胱氨酸富集区,调查显示截至目前为止,外显子3和外显子4突变超过总数的95%,>90%的UMOD突变为错义突变。文献已报道9种错义突变:c.96C>G(p.C32W);c.326T>A(p.V109E); c.553C>G(p.R185G);c.586G>A(p.D196N);c.651C>G (p.C217W);c.667T>C(p.C223R);c.707C>A (p.P236E);c.744C>G(p.C248W);c.1462G>C (p.G488R)[20-23]。UMOD基因的外显子3和外显子4发生错义突变,可影响半胱氨酸残基之间二硫键的形成,进而影响蛋白质的正常折叠,导致其在肾小管上皮细胞内质网中异常沉积并引起尿液排泄减少[24]。此外,体外实验及小鼠基因敲除实验表明尿调节素可预防尿路感染和尿路结石,并可通过与钾通道蛋白相互作用,调节其功能进而影响髓绊升支粗段肾小管的运输系统[25-26],但目前尚无研究表明尿调节素生成减少的患者出现尿路感染或尿路结石等。
ADTKD-REN以往称家族性少年型高尿酸血症肾病2型(FJHN2),FJHN2型为常染色体显性遗传的肾小管间质性疾病,肾活检病理组织学特点有:局部肾小管萎缩、继发肾小球瘢痕及肾间质纤维化,免疫染色可见肾小球球旁器颗粒细胞内肾素及肾素原减少[27]。为了突出其遗传方式及致病基因,KDIGO建议使用更简明而易于理解的名称“ADTKD-REN”,致病基因为REN定位于染色体1q32.1,编码肾素(renin),肾素是一种蛋白酶,可分解血管紧张素为血管紧张素1并调节血管紧张素1的生成。研究发现REN突变可能为编码信号肽的外显子1发生突变,现已证实4种REN突变:(p.Leu16del)、错义突变(p.Leu16Arg)、(p.Cys20Arg)、(p.Trp10Arg)[28-30],导致异常肾素在细胞内沉积,进而引起肾小球入球小动脉上肾素生成细胞凋亡,但是REN突变引起肾小管间质纤维化的机制还不清楚,此外,ADTKD-REN的其他临床表现都可能与肾素分泌减少引起的肾素-血管紧张素-醛固酮系统功能亢进相关[29]。
ADTKD-HNF1B以往称青少年起病的成人型糖尿病5型(MODY5)、肾囊肿糖尿病综合征(RCAD),MODY是1960年提出的一种常染色体显性遗传的单基因遗传性糖尿病,现已发现13种亚型MODY1-13,ADTKD-HNF1B为MODY5型,由HNF1B基因突变所致,其主要临床特点为:糖尿病、肾功能不全、胰腺畸形、泌尿生殖道畸形、肝功能异常;低镁血症及低钾血症等[1]。HNF1B基因定位于染色体17q12,含有9个外显子,编码HNF1B,可调节肾脏、胰腺以及肝脏的多基因表达过程,研究表明HNF1B基因突变有106种(http://links.lww.com/ MD/A178),其中包括HNF1B基因总体缺失(34%),错义突变(31%),移码缺失或插入(15%),无义突变(11%)以及拼接点突变(8%)[31],且HNF1B基因突变影响PKD2和SOCS3的表达[32],其中PKD2参与肾囊肿的形成,SOCS3与早期糖尿病发生相关,但是HNF1B基因突变导致肾间质纤维化的机制还有待研究。
针对上述四种遗传性间质性肾炎,KDIGO提出了新的定义——ADTKD,归纳总结了其临床特点及病理改变特点,并提出了ADTKD的可疑诊断和确诊诊断标准(表2)及基因诊断方法(表3)。

表2ADTKD诊断标准[1]
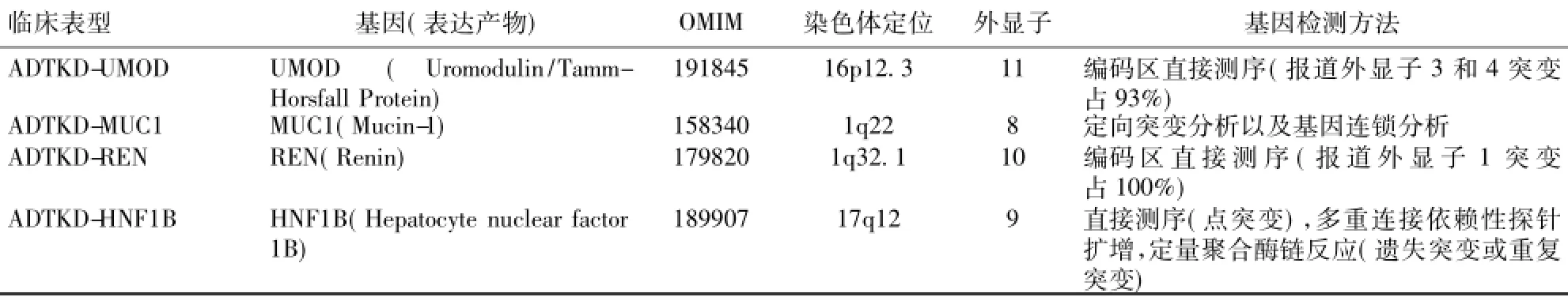
表3ADTKD致病基因突变检测[28,30,33-34]
小结:NPHP临床表现无特异性,致病基因种类较多,且只在1/3的患者中发现有致病基因突变,剩余2/3的患者致病基因还有待研究,其中检测NPHP1杂合子或纯合子基因突变最有诊断价值,但是临床分型还不够完善,容易漏诊。目前对于NPHP的治疗还比较局限,主要包括控制血压以延缓病情进展等对症治疗及适于肾功能不全患者的肾替代疗法[35]。ADTKD临床表现和病理组织学改变也无特异性,在国内相关研究较少,临床容易漏诊,迄今为止,还未有针对各型ADTKD的特异治疗,但是ADTKD的子类型唯有通过基因检测进行诊断,所以NPHP及ADTKD致病基因的相关研究进展无疑为此类疾病的诊断及治疗提供了有利条件,对基因检测、疾病预防、诊断及治疗等具有重要意义。
1Eckardt KU,AlperSL,Antignac C,etal.Autosomaldominant tubulointerstitialkidneydisease:diagnosis,classification,and management-A KDIGO consensus report.Kidney Int,2015,88(4): 676-683.
2徐峰,曾彩虹.家族性遗传性间质性肾炎.肾脏病与透析肾移植杂志,2009,18(2):167-172.
3Fanconi G,Hanhart E,von Albertini A,et al.Familial,juvenile nephronophthisis(idiopathic parenchymal contracted kidney).Helv Paediatr Acta,1951,6(1):1-49.
4Wolf MT.Nephronophthisis and related syndromes.Curr Opin Pediatr,2015,27(2):201-211.
5Gee HY,Otto EA,Hurd TW,et al.Whole-exome resequencing distinguishes cystickidneydiseases fromphenocopies inrenal ciliopathies.Kidney Int,2014,85(4):880-887.
6Mergen M,Engel C,Müller B,et al.The nephronophthisis gene product NPHP2/Inversin interacts with Aurora A and interferes with HDAC6-mediatedcilia disassembly.Nephrol Dial Transplant,2013,28 (11):2744-2753.
7Hildebrandt F,AttanasioM,OttoE.Nephronophthisis:disease mechanisms of a ciliopathy.J Am Soc Nephrol,2009,20(1):23-35.
8Leightner AC,Hommerding CJ,Peng Y,et al.The Meckel syndrome protein meckelin(TMEM67)is a key regulator of cilia function but is not required for tissue planar polarity.Hum Mol Genet,2013,22(10): 2024-2040.
9Davis EE,Zhang Q,Liu Q,et al.TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum.Nat Genet,2011,43 (3):189-196.
10 Fehrenbach H,Decker C,Eisenberger T,et al.Mutations in WDR19 encoding the intraflagellar transport component IFT144 cause a broad spectrum of ciliopathies.Pediatr Nephrol,2014,29(8):1451-1456.
11 Chaki M,Airik R,Ghosh AK,et al.Exome capture reveals ZNF423 and CEP164 mutations,linking renal ciliopathies to DNA damage response signaling.Cell,2012,150(3):533-548.
12 Hoff S,Halbritter J,Epting D,et al.ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3.Nat Genet,2013,45(8):951-956.
13 Failler M,Gee HY,Krug P,et al.Mutations of CEP83 cause infantile nephronophthisis and intellectual disability.Am J Hum Genet,2014,94(6):905-914.
14 O'Toole JF,Liu Y,Davis EE,et al.Individuals with mutations in XPNPEP3,whichencodesamitochondrialprotein,developa nephronophthisis-like nephropathy.J Clin Invest,2010,120(3): 791-802.
15 Hurd TW,Otto EA,Mishima E,et al.Mutation of the Mg2+transporter SLC41A1 results in a nephronophthisis-like phenotype.J Am Soc Nephrol,2013,24(6):967-977.
16 Bleyer AJ,Kmoch S,Antignac C,et al.Variable clinical presentation of an MUC1 mutation causing medullary cystic kidney disease type 1.Clin J Am Soc Nephrol,2014,9(3):527-535.
17 Bleyer AJ,Kmoch S.Autosomal dominant tubulointerstitial kidney disease:of names and genes.Kidney Int,2014,86(3):459-461.
18 Duncan H,Dixon AS.Gout,familial hypericaemia,and renal disease.Q J Med,1960,29:127-135.
19 Dahan K,FuchshuberA,AdamisS,etal.Familialjuvenile hyperuricemic nephropathy and autosomal dominant medullary cystic kidney disease type 2:two facets of the same disease?J Am Soc Nephrol,2001,12(11):2348-2357.
20 Williams SE,Reed AA,Galvanovskis J,et al.Uromodulin mutations causing familial juvenile hyperuricaemic nephropathy lead to protein maturation defects and retention in the endoplasmic reticulum.Hum Mol Genet,2009,18(16):2963-2974.
21 Turner JJ,Stacey JM,Harding B,et al.UROMODULIN mutations cause familial juvenile hyperuricemic nephropathy.J Clin Endocrinol Metab,2003,88(3):1398-1401.
22 Yang H,Wu C,Zhao S,et al.Identification and characterization of D8C,a novel domain present in liver-specific LZP,uromodulin and glycoprotein2,mutatedinfamilialjuvenilehyperuricaemic nephropathy.FEBS Lett,2004,578(3):236-238.
23 Liu M,Chen Y,Liang Y,et al.Novel UMOD mutations in familial juvenile hyperuricemic nephropathy lead to abnormal uromodulin intracellular trafficking.Gene,2013,531(2):363-369.
24 Joosten H,Strunk AL,Meijer S,et al.An aid to the diagnosis of genetic disorders underlying adult-onset renal failure:a literature review.Clin Nephrol,2010,73(6):454-472.
25 Rampoldi L,ScolariF,AmorosoA,etal.Therediscoveryof uromodulin(Tamm-Horsfallprotein):fromtubulointerstitial nephropathy to chronic kidney disease.Kidney Int,2011,80(4): 338-347.
26 Renigunta A,Renigunta V,Saritas T,et al.Tamm-Horsfall glycoprotein interacts with renal outer medullary potassium channel ROMK2 and regulates its function.J Biol Chem,2011,286(3):2224-2235.
27 Kmoch S, ivná M,Bleyer AJ.Familial Juvenile Hyperuricemic Nephropathy Type 2.GeneReviews [Internet].Seattle(WA): University of Washington,Seattle;1993-2015.
28 Beck BB,Trachtman H,Gitman M,et al.Autosomal dominant mutation in thesignalpeptideofrenininakindredwithanemia,hyperuricemia,and CKD.Am J Kidney Dis,2011,58(5):821-825.
29 Zivná M,Hu。lková H,Matignon M,et al.Dominant renin gene mutations associated with early-onset hyperuricemia,anemia,and chronic kidney failure.Am J Hum Genet,2009,85(2):204-213.
30 Bleyer AJ,Zivná M,Hulková H,et al.Clinical and molecular characterization of a family with a dominant renin gene mutation and response to treatment with fludrocortisone.Clin Nephrol,2010,74(6): 411-422.
31 Alvelos MI,Rodrigues M,Lobo L,et al.A novel mutation of the HNF1B gene associated with hypoplastic glomerulocystic kidney disease and neonatal renal failure:a case report and mutation update.Medicine(Baltimore),2015,94(7):e469.
32 Mancusi S,La Manna A,Bellini G,et al.HNF1B mutation affects PKD2 and SOCS3 expression causing renal cysts and diabetes in MODY5 kindred.J Nephrol,2013,26(1):207-212.
33 Kirby A,Gnirke A,Jaffe DB,et al.Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing.Nat Genet,2013,45(3):299-303.
34 Ekici AB,Hackenbeck T,Morinière V,et al.Renal fibrosis is the common featureofautosomaldominanttubulointerstitialkidney diseases caused by mutations in mucin 1 or uromodulin.Kidney Int,2014,86(3):589-599.
35 Slaats GG,Lilien MR,Giles RH.Nephronophthisis:should we target cysts or fibrosis?Pediatr Nephrol,2015:1-10
Advance in research of pathogenic genes in hereditary tubule-interstitial nephritis
QIAO Panpan,WANG Zhaohui
Department of nephrology,Shanghai Ruijin Hospital,Shanghai Jiaotong university School of medicine,Shanghai 200025,China
Recently,there has been some new research progresses about pathogenic genes of hereditary tubuleinterstitial nephritis,which has been confirmed as 20 kinds of pathogenic genes of nephronophthisis.Kidney Disease: Improving Global Outcomes(KDIGO)proposes adoption of a new terminology“Autosomal Dominant Tubulointerstitial Kidney Disease(ADTKD)”and suggests diagnostic criteria for autosomal dominant tubule-interstitial kidney disease.There has been confirmed as 4 kinds of pathogenic genes of ADTKD(MUC1,UMOD,REN,HNF1B).The study of pathogenic genes,pathologic changes and clinical symptoms has great significance for the prevention,gene diagnosis,treatment and prognosis of hereditary tubule-interstitial nephritis.
nephronophthisisautosomal dominanttubulointerstitial kidney diseasemedullary cystic kidney diseasepathogenic genes
2015-11-06
(本文编辑律舟莫非)
10.3969/cndt.j.issn.1006-298X.2016.01.013
国家重点基础研究发展计划(973计划)(2012CB5176-04);国家自然科学基金(81170634)
上海交通大学医学院附属瑞金医院肾脏科(上海,200025)
