液液萃取—气相色谱法同时测定地下水中16种有机氯农药
2016-03-13仇秀梅董学林刘亚东方晓青兰秀敏
仇秀梅 董学林 刘亚东 宋 洲 方晓青 兰秀敏
(1.湖北省地质实验测试中心,湖北 武汉 430034;2.湖北城市建设职业技术学院,湖北 武汉 430205)
有机氯农药(OCPs)是一类人工合成的高毒性、难分解、易残留有机化合物,曾作为杀虫剂在我国农业生产过程中大量使用,造成土壤中大量残留,并随着喷灌水、雨水的冲刷、淋溶、渗透等作用进入地表水或地下水中。大多数OCPs都已经被列为《关于持久性有机污染物的斯德哥尔摩公约》中首批需要控制和减少的污染物[1],如滴滴涕、氯丹、艾氏剂、狄氏剂、异狄氏剂、七氯、六氯苯等,可通过食物链富集,对生态环境和人类健康产生极大的危害。由于我国人均水资源严重不足,加上地表水污染严重,很多地区只能开发利用地下水,因此地下水中OCPs监测十分必要。
目前,水中OCPs的测定方法较多,如光学免疫法[2]、电化学酶联免疫法[3]、气相色谱(GC)法[4-6],[7]81-82、GC/质谱(MS)联用法[8-9]等。GC法因具有分离效果好、灵敏度高、分析速度快、经济等优点而备受青睐。OCPs水样的前处理方法主要有固相萃取法[10-11]、液液萃取(LLE)法[12-13]、固相微萃取法[14]等。LLE法是一种经典的方法,因成本低廉而使用率较高。在已有报道中,针对地表水、土壤、植物中OCPs测定的研究较多。在水中的OCPs测定方面,许桂苹等[15]用LLE—GC法定量测定邕江水中8种OCPs,方法检出限为0.020~0.075 μg/L,回收率为90.1%~101.2%;陆华[16]采用LLE—GC法同时测定地表水中17种OCPs,方法检出限为0.015~0.062 μg/L,回收率为72.8%~93.5%;梁素丹等[17]建立了LLE—GC法测定水体中16种OCPs的方法,检出限低至0.004~0.082 μg/L,并应用于珠海5个水库水源水中的OCPs测定。由于地下水中OCPs的污染水平相对地表水来说更低,因此对方法的检出限要求更高。
本研究进行了LLE—GC法用于同时测定地下水中16种OCPs的探索,通过优化衬管、进样口温度、载气流速、升温程序和检测器温度等条件,确定了最佳色谱条件,建立了一种快捷、高效、准确、灵敏的方法,并成功应用于实际地下水水样的测定。
1 实验部分
1.1 主要仪器与试剂
美国Agilent公司的7890B型GC仪,检测器为电子捕获检测器(ECD);瑞典Biotage公司的Turbo VapⅡ型平行蒸发仪;美国Millipore公司的Milli-Q Direct 8型纯水机。
14种OCPs(α-六六六、β-六六六、γ-六六六、δ-六六六、p,p’-滴滴伊、o,p’-滴滴涕、p,p’-滴滴滴、p,p’-滴滴涕、六氯苯、艾氏剂、七氯、环氧七氯β、狄氏剂、异狄氏剂)混合标准溶液,质量浓度为100.0 mg/L。顺式氯丹和反式氯丹混合标准溶液,质量浓度为100.0 mg/L。p,p’-滴滴涕、异狄氏剂标准溶液,质量浓度均为100.0 mg/L。替代物2,4,5,6-四氯间二甲苯标准溶液,质量浓度为500.0 mg/L。正己烷,农残级;丙酮,色谱纯;无水硫酸钠和氯化钠,分析纯,700 ℃煅烧6 h,冷却后备用。
1.2 标准溶液的配制
将14种OCPs混合标准溶液、顺式氯丹和反式氯丹混合标准溶液用正己烷稀释成质量浓度为2.0 mg/L的16种OPCs混合标准储备液。制作标准曲线时用正己烷将16种OCPs混合标准储备液稀释成质量浓度分别为1、5、10、20、40、60 μg/L的系列溶液,同时均加入质量浓度为50 μg/L的替代物。除衬管的优化过程外,其他色谱条件的优化过程所用OCPs质量浓度均为60 μg/L。衬管优化过程中,将p,p’-滴滴涕、异狄氏剂标准溶液用正己烷分别逐级稀释至100 μg/L,用于测定分解率,分解率的测定和计算方法参见文献[18]。上述所有溶液均于-18 ℃密封保存。
1.3 基本色谱条件
若无特别说明,基本色谱条件如下:色谱柱为美国Agilent公司的HP-5(30 m×0.32 mm×0.25 μm),载气为氮气,流量为0.65 mL/min;进样口温度为250 ℃;进样量为1.0 μL;进样方式为脉冲不分流进样,脉冲压力为275.8 kPa,脉冲时间为0.75 min;升温程序为从120 ℃起以10 ℃/min升至220 ℃,保持16 min,继续以10 ℃/min升至250 ℃,保持2 min;隔垫吹扫模式为标准模式,流量为3.0 mL/min;检测器温度为320 ℃;尾吹气流量为30 mL/min。
1.4 水样前处理及测定
在预先用丙酮润洗过的1 L分液漏斗中加入30 g氯化钠,将1 L水样转入分液漏斗中,用15 mL丙酮分3次润洗水样瓶内壁,一并转入分液漏斗中。加入50 mL正己烷,振荡萃取5 min,静置10~30 min后,将有机相转入250 mL锥形瓶中,再向水相中加入25 mL正己烷重复萃取2次,合并3次的有机相作为萃取液。向萃取液中加入少量无水硫酸钠以去除有机相中的水分,过滤后在45 ℃、68.9 kPa条件下用平行蒸发仪浓缩至体积小于0.5 mL,用正己烷定容到1.0 mL,用最优化的色谱条件测定。
1.5 质量控制
用超纯水配制7个含16种OCPs且加标质量浓度为5 ng/L的平行样品,按照水样前处理及测定方法进行测定,检出限用式(1)进行计算。
MLD=3.143×S
(1)
式中:MDL为检出限,ng/L;S为测定的7个平行样品的标准偏差,ng/L。
用超纯水配制含16种OCPs且质量浓度分别为5、20、60 ng/L的加标样(含替代物质量浓度为50 ng/L),按照水样前处理及测定方法进行测定,平行测定7次取平均值,并计算回收率及相对标准偏差(RSD)。
2 结果与讨论
2.1 色谱条件的优化
2.1.1 衬 管
由于异狄氏剂和p,p’-滴滴涕易在进样口分解,其分解率与衬管的选择有关,因此在使用GC法分析OCPs时,应先测定异狄氏剂和p,p’-滴滴涕的分解率。对比研究了100 μg/L的p,p’-滴滴涕、异狄氏剂标准溶液在4 mm内径中空超高惰性不分流衬管(型号为5190-2292)和4 mm内径中空惰性不分流衬管(型号为5181-3316)两种衬管下的分解率。结果表明,p,p’-滴滴涕在型号为5190-2292和5181-3316的衬管中的分解率分别为1.15%、1.03%,均满足小于15%的质量控制要求[19];异狄氏剂在型号为5181-3316的衬管中分解高达48.2%,而在型号为5190-2292的衬管中分解率仅为2.0%。因此,以下实验中均采用型号为5190-2292的4 mm内径中空超高惰性不分流衬管。
2.1.2 进样口温度
进样口温度过低,会导致目标化合物气化不完全;进样口温度过高,部分OCPs会发生分解,特别是异狄氏剂和p,p’-滴滴涕等[7]82-83。图1对比了进样口温度分别为230、250、280 ℃时,16种OCPs和替代物的响应情况。结果表明,绝大部分化合物的信号强度都在进样口温度为250 ℃时最强,优于《气相色谱法测定水中有机氯农药和多氯联苯类化合物》(SL 497—2010)的推荐温度280 ℃,因此本研究选用进样口温度为250 ℃。
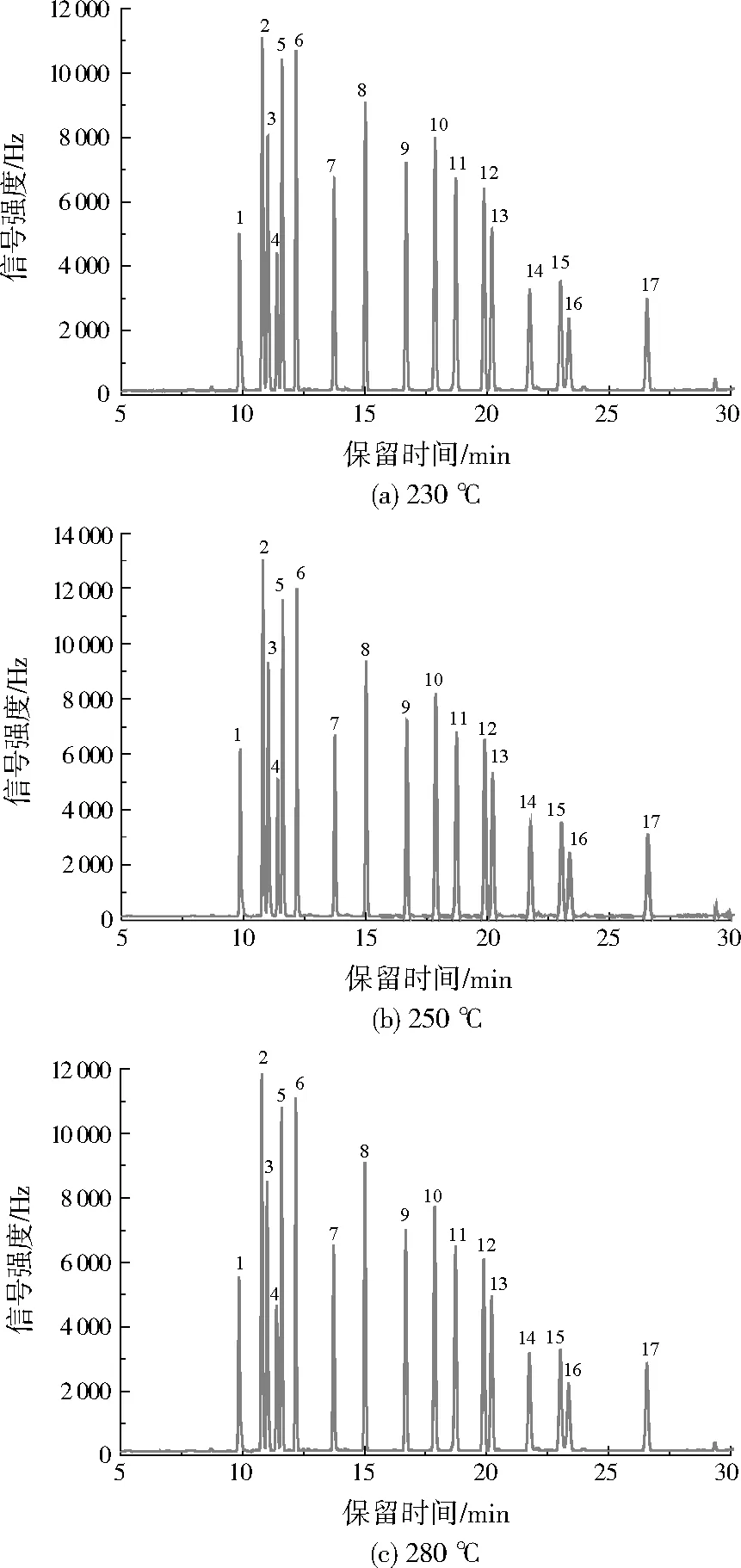
注:1—2,4,5,6-四氯间二甲苯;2—α-六六六;3—六氯苯;4—β-六六六;5—γ-六六六;6—δ-六六六;7—七氯;8—艾氏剂;9—环氧七氯B;10—反式氯丹;11—顺式氯丹;12—p,p’-滴滴伊;13—狄氏剂;14—异狄氏剂;15—p,p’-滴滴滴;16—o,p’-滴滴涕;17—p,p’-滴滴涕。图2至图5同。
图1 不同进样口温度下的色谱图
Fig.1 Chromatogram under different inlet temperature
2.1.3 载气流速
色谱柱载气流速对保留时间、峰形和分离度均有影响,考察了0.40、0.65 mL/min两种载气流速下的目标化合物分离和响应情况,结果见图2。当载气流量为0.40 mL/min时,目标化合物出峰较慢,且信号强度较低;而当载气流量为0.65 mL/min时,目标化合物具有更好的分离度,峰形也更加尖锐,信号强度高,且出峰快。为此,色谱柱的载气流速设置为0.65 mL/min。
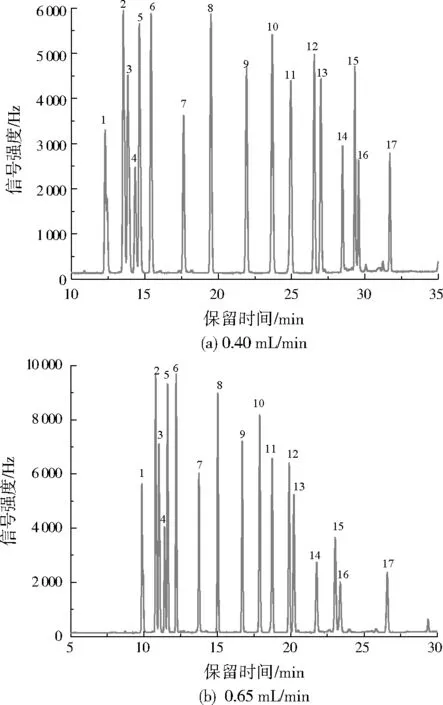
图2 不同载气流速下的色谱图Fig.2 Chromatogram under different carrier gas flow rate
2.1.4 升温程序
(1) 初始温度
SL 497—2010中推荐的色谱柱升温程序初始温度为150 ℃,本研究对比了初始温度为120、150 ℃对目标化合物分离情况的影响,结果如图3所示。由图3可见,初始温度为150 ℃时,α-六六六、六氯苯、β-六六六、γ-六六六分离效果较差,峰型也不尖锐,并且出现拖尾现象;而初始温度为120 ℃时,16种OCPs与替代物都可完全分离,峰形尖锐,信号强度也明显强于150 ℃时。因此,选择初始温度为120 ℃。
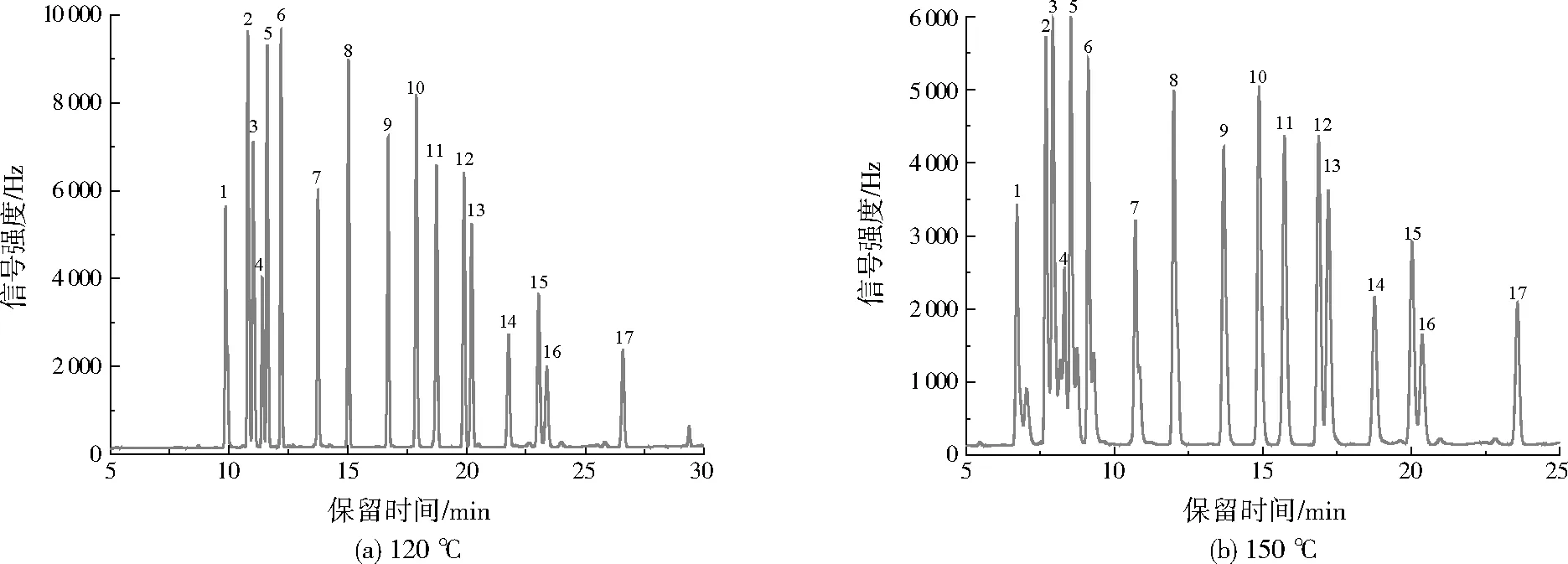
图3 不同初始温度下的色谱图Fig.3 Chromatogram under different initial temperature

图4 不同升温速率下的色谱图Fig.4 Chromatogram under different programming temperature rate
(2) 升温速率
研究了5、10、20 ℃/min 3种升温速率对目标化合物分离情况的影响,结果如图4所示。3种升温速率下大部分化合物均可以达到良好的分离效果。当升温速率为5 ℃/min时,p,p’-滴滴伊和狄氏剂的色谱峰分离效果不好,且出峰时间较晚;当升温速率为20 ℃/min时,α-六六六和六氯苯的色谱峰分离效果不好;当升温速率为10 ℃/min时,16种OCPs和替代物都能进行完全分离。因此,选择10 ℃/min作为最佳升温速率。
2.1.5 检测器温度
检测器温度应低于色谱柱的最高使用温度。由于HP-5色谱柱的最高使用温度为325 ℃,因此选择250、280、320 ℃3个检测器温度进行优化,结果如图5所示。由图5可见,大部分化合物都在检测器温度为320 ℃时信号强度最大,因此检测器温度选择320 ℃。
综上所述,优化后的最佳色谱条件为选用4 mm内径中空超高惰性不分流衬管;进样口温度250 ℃;载气流速0.65 mL/min;升温程序:从120 ℃开始以10 ℃/min升至220 ℃,保持16 min,继续以10 ℃/min升至250 ℃,保持2 min;检测器温度320 ℃。16种OCPs可以在27 min内快速、完全分离,实现同时测定。
2.2 方法性能评价

图5 不同检测器温度下的色谱图Fig.5 Chromatogram under different detection temperature

化合物保留时间/min线性回归方程1)R2检出限/(ng∙L-1)α-六六六10.876y=753.6x-15370.99811.0六氯苯11.102y=559.9x-1430.99961.2β-六六六11.486y=282.6x-1930.99971.0γ-六六六11.693y=677.8x-11370.99861.1δ-六六六12.276y=684.6x-11730.99751.5七氯13.843y=506.9x-9240.99730.9艾氏剂15.138y=631.1x-9360.99870.9环氧七氯B16.839y=547.0x-6080.99911.5反式氯丹18.021y=664.0x-8060.99901.1顺式氯丹18.881y=580.4x-5260.99950.8p,p’-滴滴伊20.047y=553.3x-9440.99701.1狄氏剂20.379y=497.5x-7480.99581.1异狄氏剂21.943y=379.3x-7170.99630.9p,p’-滴滴滴23.219y=394.3x-6050.99541.1o,p’-滴滴涕23.569y=267.7x-3800.99541.1p,p’-滴滴涕26.766y=340.3x-8500.99640.6
注:1)x为OCPs的质量浓度,μg/L;y为色谱峰面积。
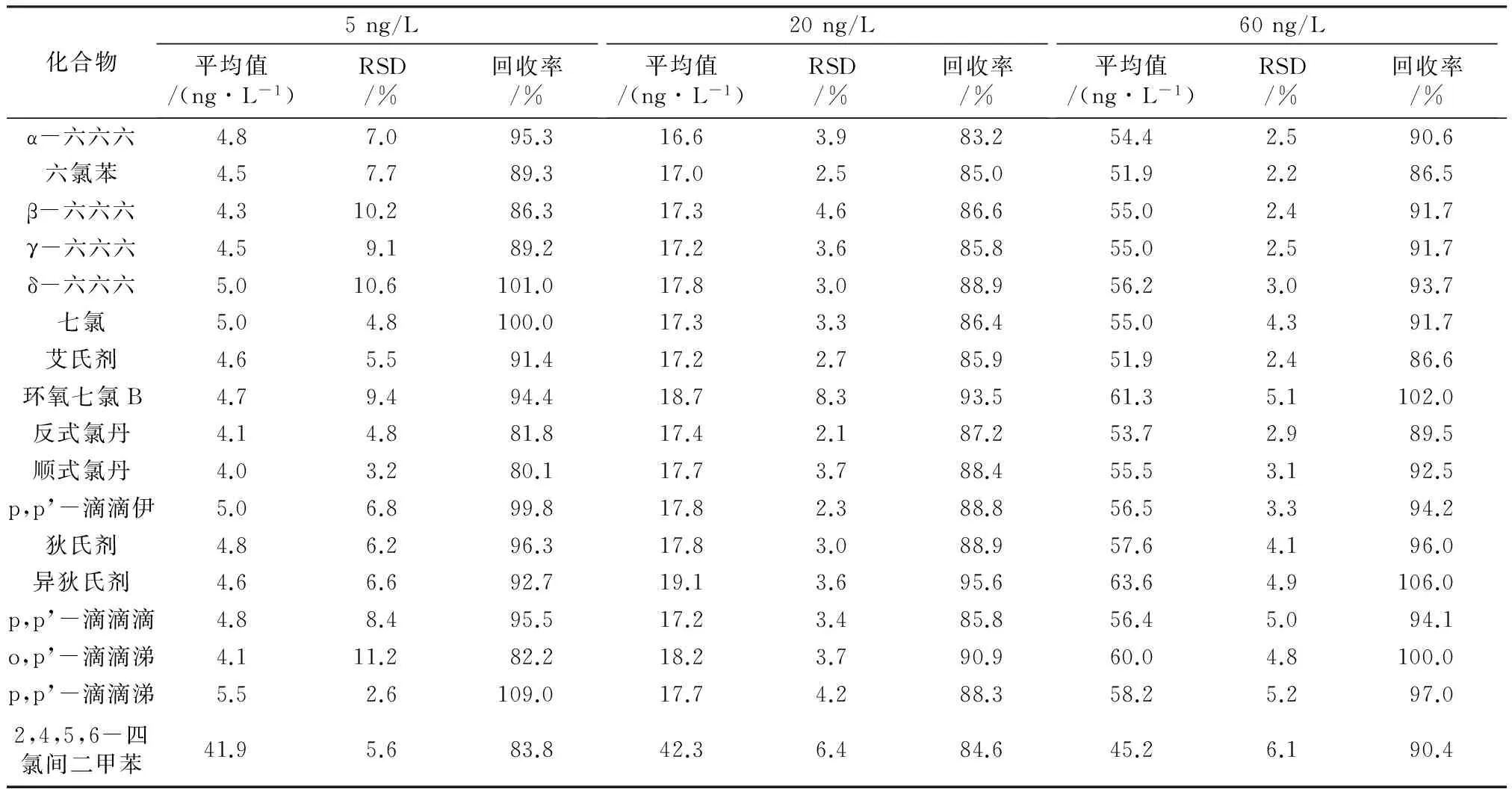
表2 准确度和精密度
2.2.1 标准曲线及检出限
在2.1节得到的最佳色谱条件下,16种OCPs的标准曲线与检出限结果见表1。16种OCPs的R2均大于0.995 0,表明各目标化合物在1~60 μg/L线性关系良好。
16种OCPs的检出限为0.6~1.5 ng/L,低于SL 497—2010的检出限水平,也明显低于文献[7]、[13]、[16]、[17]、[20]的水平,能够适用于地下水中低浓度的OCPs测定。
2.2.2 准确度和精密度
表2显示,16种OCPs的低(5 ng/L)、中(20 ng/L)、高(60 ng/L)3个浓度的回收率达到80.1%~109.0%,RSD为2.1%~11.2%,方法的准确度和精密度均满足SL 497—2010的质量控制要求。替代物2,4,5,6-四氯间二甲苯的回收率达到了83.8%~90.4%,RSD为5.6%~6.4%。
3 实际水样的测定
在湖北某地下水采集了3个水样,分别为样品1、样品2和样品3,按照1.4节的方法进行测定,结果见表3。由表3可见,样品2中α-六六六被检出,检出质量浓度为7.1 ng/L;样品3中β-六六六和δ-六六六被检出,检出质量浓度分别为5.2、8.1 ng/L。由此可见,本研究建立的方法可以应用于实际地下水的测定。

表3 实际水样测定结果1)
注:1)nd表示未检出。
4 结 论
采用正己烷液液萃取,联合GC法建立了一种准确、灵敏的地下水中16种OCPs的快速测定方法。优化后的最佳色谱条件为选用4 mm内径中空超高惰性不分流衬管;进样口温度250 ℃;载气流速0.65 mL/min;升温程序:从120 ℃开始以10 ℃/min升至220 ℃,保持16 min,继续以10 ℃/min升至250 ℃,保持2 min;检测器温度320 ℃。16种OCPs在1~60 μg/L线性关系良好,R2>0.995 0,检出限为0.6~1.5 ng/L,回收率达到80.1%~109.0%,RSD为2.1%~11.2%,符合SL 497—2010的质量控制要求,并能满足实际地下水水样的测定。
[1] 马晗宇,刘菲,刘玉龙.气相色谱法测定地下水中有机氯农药和多氯联苯[J].岩矿测试,2010,29(5):527-530.
[2] MAURIZ E,CALLE A,MANCLS J J,et al.Optical immunosensor for fast and sensitive detection of DDT and related compounds in river water sample[J].Biosensors and Bioelectronics,2007,22(7):1410-1418.
[3] VALENTINI F,COMPAGNONE D,GIRAUDI G,et al.Electrochemical ELISA for the screening of DDT related compounds:analysis in waste waters[J].Analytica Chimica Acta,2003,487(1):83-90.
[4] DAI Guohua,LIU Xinhui,LIANG Gang,et al.Distribution of organochlorine pesticides (OCPs) and polychlorinated biphenyls (PCBs) in surface water and sediments from Baiyangdian Lake in North China[J].Journal of Environmental Sciences,2011,23(10):1640-1649.
[5] 郭晓辰,饶竹,高冉.气相色谱法测定地下水中拟除虫菊酯有机氯百菌清等24种农药残留[J].岩矿测试,2014,33(3):406-412.
[6] 王娜.液-液萃取富集—气相色谱法测定水中有机氯农药和多氯联苯[J].理化检验(化学分册),2011,47(3):327-330.
[7] 黄玉娟,吴春发,罗飞,等.气相色谱法检测地下水中六六六和滴滴涕[J].环境监测管理与技术,2011,23(3).
[8] 陈军,张宗祥,杨文武,等.固相萃取—气质联用法测定水中有机氯农药和氯苯类化合物[J].环境监测管理与技术,2015,27(1):46-49.
[9] 李江,徐特秀.水中34种有机氯农药和氯苯类化合物测定的固相萃取—气相色谱—质谱分析法[J].污染防治技术,2015,28(3):56-61.
[10] 刘静,曾兴宇,烟卫.固相萃取气相色谱法测定水中氯苯类化合物和有机氯农药[J].环境化学,2010,29(5):980-981.
[11] 许秀艳,张颖,程麟钧,等.固相萃取—GC/MS法测定水中16种有机氯农药[J].环境监测管理与技术,2010,22(6):51-54.
[12] 江树广.气相色谱法同时测六六六、滴滴涕、六氯苯、七氯、百菌清[J].中国给水排水,2011,27(18):92-95.
[13] 王美飞,杨丽莉,胡恩宇,等.液-液萃取—气相色谱法同时测定地表水中17种硝基苯类和氯苯类化合物[J].中国环境监测,2011,27(增刊):19-23.
[14] 邰超,齐永安,庞玉娟,等.固相微萃取—气相色谱法测定水中痕量有机氯农药[J].环境监测管理与技术,2007,19(5):26-29.
[15] 许桂苹,蒋建宏,白海强,等.液液萃取—气相色谱法测定水中有机氯的方法研究[J].安徽农业科学,2011,39(14):8564-8566.
[16] 陆华.液液萃取—气相色谱法测定地表水中17种有机氯农药研究[J].环境科学与管理,2014,39(2):94-96.
[17] 梁素丹,陈剑刚,白艳玲,等.液液萃取—气相色谱法同时测定水中氯苯类化合物和有机氯农药[J].中国卫生检验杂志,2013,23(6):1385-1388.
[18] 汪雨,支辛辛,张玲金,等.C18固相膜萃取—气相色谱法测定饮用水中12种有机氯农药[J].岩矿测试,2006,25(4):301-305.
[19] Method 8081B,Organochlorine pesticides by gas chromatography[S].
[20] 乔晓平,孙彬彬.液液萃取—毛细管柱气相色谱法测定水中有机氯农药的研究[J].污染防治技术,2015,28(4):39-42.
