2-苄基—苯并咪唑与反丁烯二酸的共晶及其溶解度测定
2016-03-04王弯
王 弯
(咸宁市实验外国语学校,湖北 咸宁 437100)
2-苄基—苯并咪唑与反丁烯二酸的共晶及其溶解度测定
王 弯
(咸宁市实验外国语学校,湖北 咸宁 437100)
药物共晶是药物活性成分与共晶试剂通过分子间作用力( 如氢键) 而形成的一种新晶型,它可改善药物活性成分的理化性质和生物利用度,所以近年来有关药物共晶的研究已成为药学领域一大热点。本文主要是合成2-苄基—苯并咪唑后研究2-苄基—苯并咪唑—反丁烯二酸共晶的合成及测定其溶解度。
药物共晶;2-苄基—苯并咪唑;反丁烯二酸;合成
苯并咪唑即为咪唑的苯并衍生物。苯并咪唑衍生物是一种含氮杂环化合物,这类化合物具有抗癌、抗糖尿病和肥胖、抗菌、抗 HIV、抗氧化、抗溃疡的作用,除此之外还可以作为质子泵抑制剂、酶抑制剂、降压、脑渗透剂和 CB2 受体激动剂。苯并咪唑类化合物具有非常强的生物活性,所以苯并咪唑结构可以作为药物开发中的药效团,同时也可以作为非常多重要有机化合物合成的中间体。许多药物的产生是人们对苯并咪唑骨架优化的种类繁多的取代物所导致的,如可以作为质子泵抑制剂的奥美拉唑、兰索拉唑、泮托拉唑还有许多优秀的化合物被用于其它治疗领域中[1]。
2-苄基—苯并咪唑(结构图如图1所示)作为苯并咪唑类化合物中的一种,其结构中咪唑环上的两个氮原子具有多种生物活性。2-苄基—苯并咪唑可用于治疗轻度高血压、脑血管痉挛、胃肠平滑肌痉挛等等,但由于2-苄基—苯并咪唑在人体内溶解度相对较小,人体难以吸收而造成治疗效果不佳。因此,如何增大2-苄基—苯并咪唑的溶解度使其充分发挥治疗作用成为研究的对象。现在市面上的地巴唑片就是2-苄基—苯并咪唑的盐酸盐,制成盐酸盐后的2-苄基—苯并咪唑在人体内溶解度增加,便于人体对药物的吸收从而达到良好的治疗效果。但是研究表明,人体若摄入过量的氯离子会引起高血压、高氯血酸中毒等症状[2],因此2-苄基—苯并咪唑的盐酸盐类药物的潜在危害比较大。
本文为解决这一问题,努力寻找增加2-苄基—苯并咪唑溶解度的方法。药物共晶是增加药物溶解度一种非常重要的手段,除了可以增加药物的溶解度之外,药物共晶也是用于改善药物的物理化学性质重要方法。因此本文采用药物共晶来增加2-苄基—苯并咪唑的溶解度。

图1 2-苄基苯并咪唑结构示意图

图2 反丁烯二酸结构示意图
本文选择反丁烯二酸与2-苄基—苯并咪唑共晶,除了因为反丁烯二酸具有以上优良性质之外,还有一个重要原因。众所周知,人体酸碱是否平衡决定了一个人的健康状态,在酸碱失衡的状态下会诱发各种疾病。凡食物中所含的氯、硫、磷元素较多时,在体内代谢产物为酸性,为人体健康带来隐患。现在市面上各种药物琳琅满目,若代谢产物为酸性,将会对人体造成更大伤害,反丁烯二酸是一种弱酸,相对于盐酸这种强酸对人体的伤害小得多。
药物活性成分(Active Pharmaceutical Ingredients,APIs)的物理化学性质在很大程度上决定了药物疗效,API组分不同的剂型、型态都会使其具有不同的理化性质从而影响药效。那么在药物合成开发阶段,研究者通常根据药物本身具有的性质以及目标药物所要达到的要求来决定药物的固体型态,从而使药效最佳。确定和选择药物的最佳固体型态在科学研究和临床医学中具有十分重要的作用[3]。对于共晶,目前广泛被接受的定义是:API分子与其他生理上可接受的酸、碱、盐、非离子化合物分子以氢键、π-π堆积作用、范德华力和其他非共价键相连而结合在同一晶格中称为共晶。药物共晶任意组分在室温条件下都是固体,其中至少有一个是分子或离子型的API。正如前文所说,药物的固体型态可以通过共晶来改变,因此可以通过共晶来改变药物的晶型从而扩大药物的专利保护范围。
早在 1844年 和 1893 年就有关于醌氢醌的报道[4]。尽管如此,关于共晶的广义和狭义定义仍然是一个存在许多争议的问题。
那么,如何设计药物共晶呢?共结晶实验设计主要考虑氢键成键原理和评估潜在的分子间相互作用的强度。氢键良好的内在稳定性和方向性使之在共晶设计中具有很重要的应用。
对共晶的系统研究表明,一个良好的氢键供体可与一个良好的氢键受体按照预先设计的结构,产生相互间的分子作用[5]。
羧酸是晶体工程学研究最多的一种官能团之一[6],从形成氢键角度来看,羧酸既可以作为氢键受体又可以作为氢键給体。图3给出的是羧酸常见的氢键类型。
本文所选择的与2-苄基—苯并咪唑共晶的物质就是含有两个羧基的反丁烯二酸。
药物共晶有着非常广泛的应用,主要有以下三种:

图3 羧酸常见的几种氢键类型
改善药物的溶解度
对于水溶性较差的药物,成盐或成络合物以及将液体直接装进胶囊等是增加其水溶性的常用方法。 Childs[7]等运用晶体工程学方法,合理的设计了盐酸氟西汀与苯甲酸、琥珀酸、富马酸的共晶,并且与 API 盐酸氟西汀分别进行了粉末溶出速率实验和特性溶出速率实验对比。最终的实验结果表明:在 20°C 的粉末溶出速率实验中,琥珀酸良好的溶解性使盐酸氟西汀-琥珀酸共晶溶解后即分解成了盐酸氟西汀,而苯甲酸和富马酸与盐酸氟西汀形成的共晶在实验条件下其溶解度均比盐酸氟西汀的要高。而在 10°C 的特征溶出速率实验中,盐酸氟西汀的溶出速率比苯甲酸和富马酸的共晶溶出速率要快,但是盐酸氟西汀的溶出速率远远比不上琥珀酸共晶的溶出速率。从研究结果看,只要共晶配体选择得当,药物的溶解度同样能得到有效的改善。
提高药物的稳定性
某些容易与水作用发生水合或降解的药物合成共晶之后其稳定性就极大地提高了。例如,茶碱是一种可以抗哮喘的物质,在制药过程中,茶碱与水作用后的一水合物和无水合物会发生互变,这种互变对药物质量的控制极其不利。Trask等人通过将茶碱制成共晶,改善了这种不利。茶碱在RH=98%的条件下,在一天之内就会发生解离形成水合物,而戊二酸与茶碱形成的共晶则可以稳定1天,而乙二酸与茶碱形成的共晶发生解离形成茶碱水合物的时间是7周,大大延长了解离时间[8]。
提高药物的生物利用度
生物利用度(bioavailability)是指药物被机体吸收进入循环的相对量和速率,用F表示,F=(A/D)×100%,A为进入人体循环的量,D为口服剂量。生物利用度是药物制剂质量的一个重要指标,药物的生物利用度与药物的稳定性、溶解度和溶出速率有密切的关系,药物制备成共晶以后,使得API的生物利用度有很大方面的提高[9]。
现在制备药物共晶的方法已有非常多种,主要有以下3种:
(1) 溶液合成法
溶液合成法包括溶液结晶法和超声法,本文主要介绍溶液结晶法。
溶液结晶法是比较常用的溶液合成法,其主要方法就是按照一定的化学计量比将同为流体状态的API和CCF加入到适当的溶剂中进行共结晶(包括蒸发结晶和冷却结晶)。在一个多组分的溶液中,当 API和 CCF 分子结构中含有可以形成比其他分子间作用力强的氢键的官能团,那么这种制备共晶的方法具有热力学优势[10]。
(2) 固体合成法
固体合成法主要有:升华法、研磨法、熔融法等。本文主要介绍研磨法。研磨法就是在外界机械的作用力下使固体分子之间产生分子间作用力或氢键而形成共晶,是不同固体形态之间互变的常用手段。研磨法主要有两种,一是无液干磨法(Neat Grinding,NG),二是加液共磨法(Solvent-Drop Grinding,SDG)。
NG 法也称固体研磨法。此法就是将API和CCF混合后不加入任何溶剂在研钵或球磨机中研磨,利用两种固体分子之间结构的互补性及移动性来形成共晶。此法无需溶剂,副产物少,并且绿色环保[10]。
SDG也是研磨,但与NG法不同之处在于研磨前后需加入溶剂以促进共晶的形成,此法可加速固体分子间的移动,降低体系的玻璃化转变温度Tg,从而加快共晶的生成速率[10]。
(3) 超临界流体法
此法是使 API 和 CCF在介质超临界 CO2作用下溶解而后二者之间产生分子间作用力,然后减压使体系过饱和析出共晶,这种方法需要物质在超临界 CO2中有足够的溶解度[10]。
药物共晶的形成离不开分子间作用力,其中氢键和π-π共轭作用是最常见的两种。
1)π-π共轭作用
π-π共轭作用力在不同环境下就会有不同的形成机理,同时作用力强度也会随环境的改变而发生变化。1990 年,Hunter 和 Sanders通过对边对面的 T 型堆积、错位的平行堆积、面对面式的π-π堆积这三种最典型的芳环堆积的分析就 π-π 共轭作用的本质提出了一套理论模[11],他们认为π-π共轭作用的本质是静电引力。这三种芳环堆积中最普遍的形式:错位的平行堆积这种作用力是药物共晶结构单元空间堆积的基础。
2)氢键
在分子中,与半径很小且电负性较大的 O、F、N 等以共价键结合的氢原子 X-H因极化效应具有很大的偶极矩,成键电子云将会向电负性较大的重原子核偏移,所以氢原子核周围电子分布很少,氢核外侧裸露带有很强的正电性,若在一定距离的范围内存在含有孤对电子并带有负电荷的原子,在距离和方向适中的条件下正电性的氢核与负电荷的原子就会发生静电吸引力,这种作用力就是氢键( X-H…Y,其中 X,Y 代表 F、O、N、S 等电负性大且半径小的原子)。若Y与X(Y与X可以是同种元素,也可是不同元素)存在于同一分子中,则称为分子内氢键,若存在于不同分子中,则称为分子间氢键。在氢键中,氢原子处于以共价键相互作用的原子和以静电吸引作用的原子两个强电负性原子之间,起到桥梁的作用。
一、实验
1.2-苄基—苯并咪唑的合成
(1)实验试剂
邻苯二胺(分析纯,上海安诺芳脑化学品有限公司)、苯乙酸、乙二醇、无水乙醇
(2)合成路线

(3)实验步骤
室温下,将0.541g邻苯二胺粉末(白色)与0.681g苯乙酸(无色晶体)溶于乙二醇溶液中,加热回流6 h,反应体系颜色为红色,停止反应并用冰水冷却,得到红色粗产物。用少量冷无水乙醇洗涤,室温干燥,重结晶得到微红色2-苄基—苯并咪唑。
(4)实验结果
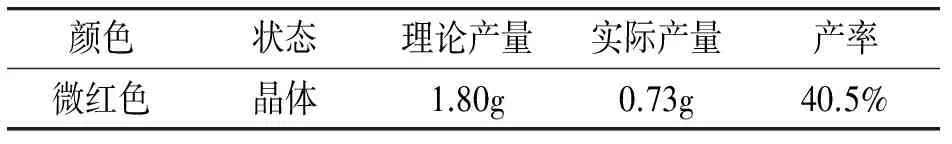
颜色状态理论产量实际产量产率微红色晶体1.80g0.73g40.5%
2.苄基—苯并咪唑与反丁烯二酸共晶
(1)实验仪器
超声波清洗机、Bruker Smart CCD衍射仪
(2)实验试剂
2-苄基—苯并咪唑(分析纯,上海安诺芳胺化学品有限公司)、反丁烯二酸、甲醇、蒸馏水
(3)实验步骤
用分析天平准确称取0.104g 2-苄基—苯并咪唑于50mL干燥小烧杯中,再准确称取0.058g反丁烯二酸(n(2-苄基—苯并咪唑):n(反丁烯二酸)=1:1)一并加入小烧杯,向小烧杯中加入10mL甲醇,用超声波清洗机振荡至固体完全溶解。静置待溶剂完全挥发,得到晶体。称重计算产量。
3.2-苄基—苯并咪唑—反丁烯二酸晶体结构晶体结构分析
(1)仪器和方法

(2)2-苄基—苯并咪唑—反丁烯二酸晶体结构分析

表1 2-苄基—苯并咪唑—反丁烯二酸晶体数据及结构精修数据

图4 2-苄基—苯并咪唑—反丁烯二酸晶体结构图

图5 2-苄基—苯并咪唑—反丁烯二酸晶体堆积图

图6 2-苄基—苯并咪唑—反丁烯二酸晶体结构中芳环的π-π堆积作用

C(1)-N(1)1.3330(15)C(12)-C(13)1.3808(19)C(1)-N(2)1.3341(15)C(12)-H(12)0.9500C(1)-C(8)1.4884(17)C(13)-C(14)1.3794(18)C(2)-C(3)1.3858(17)C(13)-H(13)0.9500C(2)-N(2)1.3864(15)C(14)-H(14)0.9500C(2)-C(7)1.3943(15)C(15)-O(2)1.2538(14)C(3)-C(4)1.3751(17)C(15)-O(1)1.2553(14)C(3)-H(3)0.9500C(15)-C(16)1.5024(16)C(4)-C(5)1.4018(18)C(16)-C(17)1.3226(17)C(4)-H(4)0.9500C(16)-H(16)0.9500C(5)-C(6)1.3784(19)C(17)-C(18)1.4817(16)C(5)-H(5)0.9500C(17)-H(17)0.9500C(6)-C(7)1.3938(16)C(18)-O(3)1.2179(14)C(6)-H(6)0.9500C(18)-O(4)1.3144(14)C(7)-N(1)1.3852(16)N(1)-O(1)2.7243(12)C(8)-C(9)1.5154(17)N(1)-H(1)0.872(14)C(8)-H(8A)0.9900N(2)-H(2)0.945(13)C(8)-H(8B)0.9900O(1)-O(5)2.6922(13)C(9)-C(10)1.3867(17)O(3)-O(6)2.8208(13)C(9)-C(14)1.3901(18)O(4)-H(4A)0.939(15)C(10)-C(11)1.386(2)O(5)-H(5A)0.920(17)C(10)-H(10)0.9500O(5)-H(5B)0.884(16)C(11)-C(12)1.375(2)O(6)-H(6A)0.917(18)C(11)-H(11)0.9500O(6)-H(6B)0.835(19)

表3 2-苄基—苯并咪唑—反丁烯二酸晶体的键角

表4 氢键键长及键角
4.2-苄基—苯并咪唑—反丁烯二酸共晶溶解度的测定
(1)验仪器
光电二极管阵列紫外-可见分光光度计
(2)验步骤
1)准确称量3.0mg 2-苄基—苯并咪唑—反丁烯二酸共晶于50mL小烧杯中,加入适量蒸馏水完全溶解,转移至50.00mL的容量瓶中。用移液管分别移取5mL、6mL、10mL、15mL于50.00mL容量瓶中,蒸馏水定容后放置待用。
2)在50mL小烧杯中加入适量水后不断加入少量2-苄基—苯并咪唑—反丁烯二酸直至2-苄基—苯并咪唑—反丁烯二酸共晶不再溶解。过滤,滤去多余的2-苄基—苯并咪唑—反丁烯二酸。
3)取出1mL饱和2-苄基—苯并咪唑—反丁烯二酸溶液稀释至50mL容量瓶中记为溶液1,再从容量瓶中取出10mL溶液于25.00mL容量瓶中记为溶液2。
4)用光电二极管阵列紫外-可见分光光度计分别测定以上溶液的吸光度。
(3)实验数据

图7 不同浓度2-苄基—苯并咪唑—反丁烯二酸共晶的吸度曲线
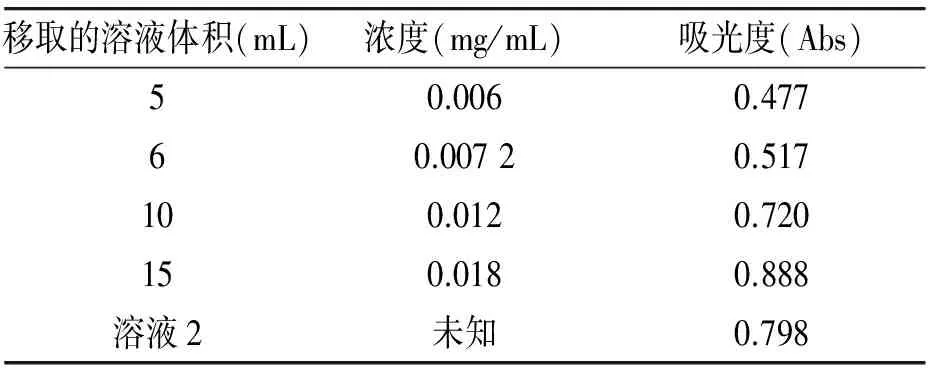
移取的溶液体积(mL)浓度(mg/mL)吸光度(Abs)50.0060.47760.00720.517100.0120.720150.0180.888溶液2未知0.798
根据以上实验数据,作出吸光度与浓度工作曲线,再根据工作曲线计算出饱和溶液的浓度,进而推算出2-苄基—苯并咪唑—反丁烯二酸共晶的溶解度。
二、结果分析
从图7可以看出2-苄基—苯并咪唑—反丁烯二酸共晶在波长213nm及270nm附近有较强的吸收,根据以上实验数据作出工作曲线为y=34.798x + 0.274 7,根据工作曲线很容易得到饱和溶液的浓度为1.88mg/mL(即为0.005 2mol/L)。查阅CA文献,得知2-苄基苯并咪唑的溶解度0.001 1mol/L,溶解度增加了接近5倍。因此通过药物共晶的方法来增加2-苄基—苯并咪唑这种药物的溶解度是可行的。这个结论为日后医学研究2-苄基—苯并咪唑这种药物奠定了一定的基础。
三、结论
1.以邻苯二胺和苯乙酸为原料合成了2-苄基—苯并咪唑

3.利用光电二极管阵列紫外-可见分光光度计间接测得2-苄基—苯并咪唑—反丁烯二酸晶体的溶解度。发现共晶后2-苄基苯并咪唑药物的溶解度大幅度增加。
[1]李英俊,张经京.苯并咪唑类化合物生物活性的研究进展[J].化学工程与装备,2013,(8):175~177.
[2]李玉贤,万焱,尚遂存.浅议盐酸盐类药物的潜在危害[J].现代预防医学,2011,(38):799~800.
[3]Datta S, Grant D. J. W. Crystal Structures of Drugs: Advances in Determination, Prediction and Engineering. Nat. ReV[J]. Drug DiscoVery 2004, (3): 42~57.
[4]Vishweshw AR P, Mcmahon JA, Bis JA,et a1. Pharmaeeutieal cocrystals[J].Pharm Sci, 2006,95(3):499~516.
[5] Remenar JF, Morissette SL, Peterson ML,et a1.Crystal engineering of novel eoerystals of a triazole dru g with 1,4-dicarboxylic acids[J]. J Am Chem Soc, 2003, 125(28):8456~8457.
[6]Kuduva SS, Craig DC, Nangia A, et a1.Cubaneearboxylic acids.crystal engineering considerations and the role of C-H?O hydrogen bonds in determining O-H?O networks[J]. J Am Chem Soc, 1999, 121(9):1936~1944.
[7] Childs SL,Chyall LJ,Dunlap JT,et a1.Crystal engineering approach to forming eoerystals of amine hydroehlorides withorganic acids.Molecular complexes of fluoxetine hydrochloridewith benzoic,suceinic,and fumaric acids[J]. Am Chem Soc, 2004, 126(41): 13 335 ~ 13 342.
[8]Tasak AV, Motherwell WD, Jones W. Physical stability enhancement of theophyine via eoerystallization[J]. Int J Pharm, 2006, 320 (1/2):114~123.
[9]弋东旭,洪鸣凰,徐军,等. 药物共晶研究进展及应用[J]. 中国抗生素杂志,2011,(8):561~565,575.
[10]张晓明,卢晓娥,李静.药物共晶制备及表征方法研究进展[J].广东化工,2013,(7):79~80.
[11]M. Sinnokrot, C. Sherrill, J. Am. Chem[J]. Soc,2004,(126): 7690.
2095-4654(2016)12-0096-06
2016-10-12
R914
A
