KDNP与KDNP·H2O的量子化学研究
2016-02-20张诗欣盛涤伦朱雅红倪德斌史胜楠庞丛丛
张诗欣,盛涤伦, 朱雅红,倪德斌,史胜楠,庞丛丛
(陕西应用物理化学研究所,陕西 西安,710061)
KDNP与KDNP·H2O的量子化学研究
张诗欣,盛涤伦, 朱雅红,倪德斌,史胜楠,庞丛丛
(陕西应用物理化学研究所,陕西 西安,710061)
以Materials Studio软件为基础,使用Dmol3模块以B3LYP/DNP方法对7-羟基-4,6-二硝基-5-氢-苯并呋咱钾(KDNP)和7-羟基-4,6-二硝基-5-氢-苯并呋咱钾的一水化物(KDNP·H2O)进行了分子性质模拟预测,并对KDNP和KDNP·H2O晶体的爆轰性质进行了模拟计算。计算结果表明KDNP分子结构具有共面性,而KDNP·H2O中H2O分子与苯并氧化呋咱环异面,二面角为29.35°;键级计算推断出两种分子均具有一定共轭性。静电势预测出KDNP· H2O比KDNP有较高的撞击感度。计算所得前线轨道能量差KDNP·H2O比KDNP小。根据以上计算结果使用最易跃迁法预测出KDNP·H2O较KDNP敏感。此外,KDNP·H2O晶体的爆轰性能与KDNP不带结晶水的晶体基本处于同一水平。
KDNP;KDNP·H2O;量子化学;感度;爆轰性质
7-羟基-4,6-二硝基-5-氢-苯并呋咱钾(KDNP)是近年来发展起来的一种性能优于斯蒂芬酸铅的无铅、无毒且环境友好的起爆药。KDNP的撞击感度和静电感度均低于常规斯蒂芬酸铅(N-LS),合成制备的KDNP·H2O在撞击感度、静电感度以及热丝感度方面也优于N-LS[1]。因此,KDNP与KDNP·H2O均为优良的新型起爆药。但是目前对其性质仍然缺乏一定的理论解释,国内外尚未见相关的量子化学研究报道。因此本研究对KDNP和KDNP·H2O进行了量子化学模型理论研究,在B3LYP/DNP水平下对KDNP和KDNP·H2O进行了SCF计算,首次给出了其全优化几何构型、电荷分布、键级、静电势和前线轨道分布,并对计算结果进行了分析和讨论。
本研究首先运用美国Accelrys 公司的Materials Studio 软件包的Dmol3 模块(密度泛函理论)B3LYP/DNP 计算KDNP与KDNP·H2O的气态分子结构和性质,获得能量最低的分子几何优化构型、电荷分布及静电势等若干量化参数;其次以GGA/PBE方法预测了两种化合物晶体状态下的爆热、爆压、爆速。
分子的初始构型由Materials Studio 软件包的Visualizer 模块获得,以Dmol3模块中的B3LYP/DNP方法对分子进行几何构型优化和频率计算,收敛精度取程序内定值,所得结构均为势能面上的极小点(无虚频),得到所计算分子的有关电荷分布、静电势、HOMO-LUMO轨道;晶体构型来自CCDC(Cambridge Crystallographic Data Centre )数据库以及实测单晶。
2 结果讨论
2.1 分子几何构型
图1为KDNP以及KDNP·H2O分子优化几何构型示意图。
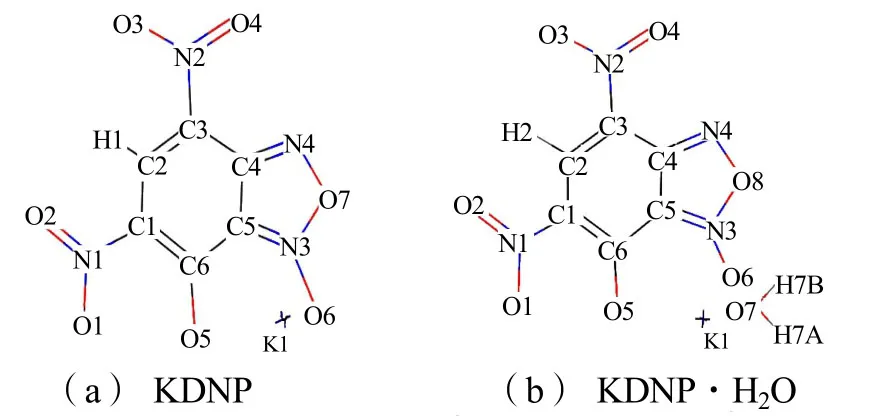
图1 KDNP与KDNP·H2O的分子优化几何构型Fig.1 The optimized geometry of KDNP and KDNP·H2O
对KDNP的构型优化表明,苯并氧化呋咱环原子之间以及环原子与钾原子之间的所有二面角均接近0°或180°,即不仅苯并氧化呋咱环具有平面结构,而且钾原子也与环共面,整个配合物的共面性较高;而KDNP·H2O的优化构型则表明苯并氧化呋咱环原子与钾原子共平面,而水分子处于另一平面,二面角为29.35°。表1 为KDNP以及KDNP·H2O分子优化几何键长。表2 为KDNP以及KDNP·H2O分子优化几何键角。
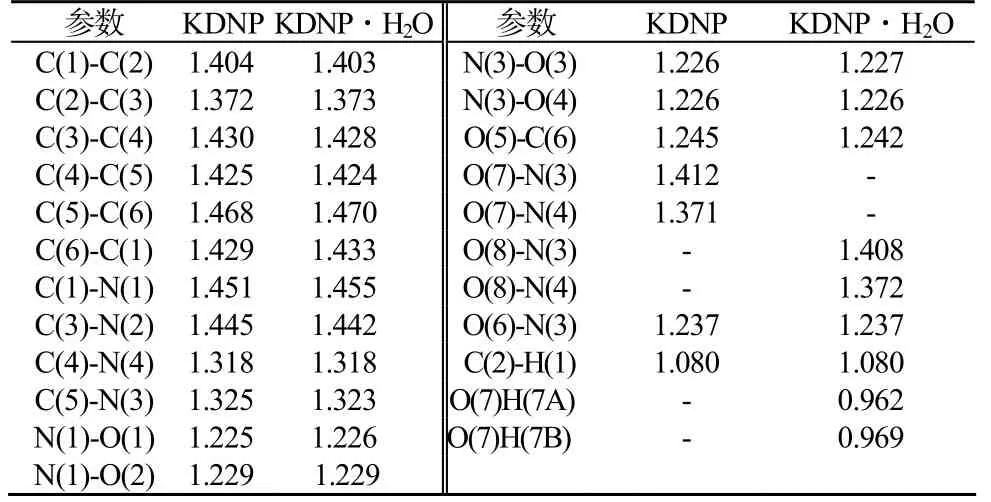
表1 B3LYP/DNP优化几何键长(Å)Tab.1 B3LYP / DNP optimized geometric bond length
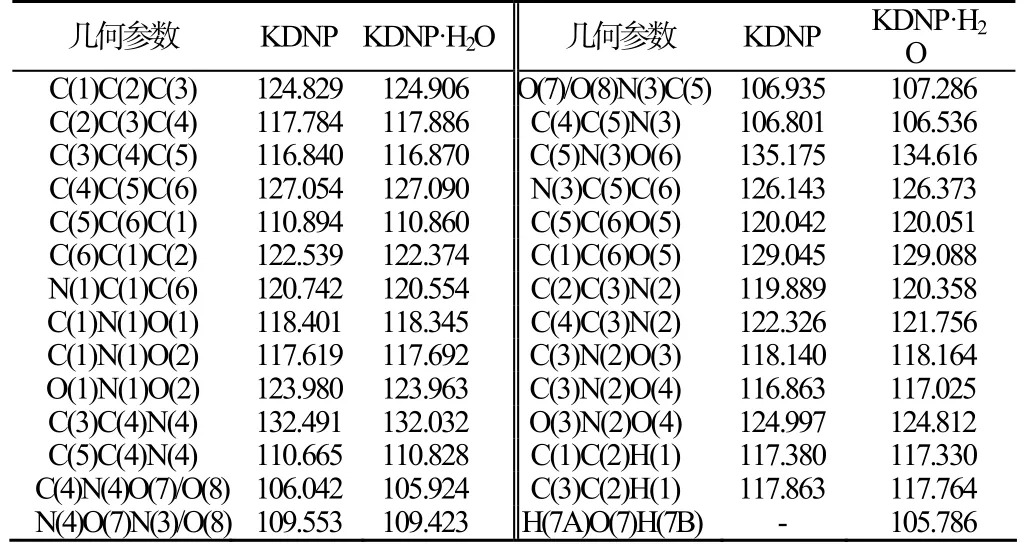
表2 B3LYP/DNP 优化几何键角 (°)Tab.2 B3LYP / DNP geometry optimization bond angle
分析表2数据可知:KDNP与KDNP·H2O的六元环骨架原子形成的键角均在111~128°变化,键角偏离120°,表明该六元环骨架内存在一定的张力。由于双键包含的电子较多,斥力较单键大,结果使分子内包含多重键的夹角增大,单键间的夹角变小,即C(4)C(5)C(6)、C(1)C(2)C(3)、C(6)C(1)C(2)键角的增大,C(3)C(4)C(5)键角的减小。而C(5)C(6)C(1) 键角的减小则是由于C(5)-C(6)、C(3)-N(3)的双键形成共轭结构,电子云部分重叠,在环张力的作用下减小(C(2)C(3)C(4)键角减小也是由于类似的原因);KDNP与KDNP·H2O的呋咱环骨架原子形成的键角均在105~111°内变化,键角偏离120°(正常sp2杂化)而接近于正五边形的内角108°。这表明呋咱环内存在一定张力,同样由单双键的原因使得键角增大和减小。由此分析,KDNP与KDNP·H2O分子内存在张力,均不稳定。
2.2 原子上净电荷和键级
表3为B3LYP/DNP水平下原子上的净电荷。由表3可见,两种分子结构中六元环中碳原子上的电荷几近为零,仅C(6)因与参与成键的O相邻而有较多的正电荷。硝基中的O的电荷均小于O(5)所带电荷,而负电荷越多,亲核性越强,与金属位点成键的能力也越强,因此O(5)是该化合物的主要配体。O(6)的电荷较多可能也是因为与K原子距离相近的缘故。同时,KDNP中K的电荷为0.904,KDNP·H2O中K的电荷为0.893,均小于+1,说明形成配位化合物时,电子从配体向金属原子转移,且KDNP·H2O转移的较多。KDNP·H2O中H2O分子的H原子、O原子的电荷为0.288、0.322,-0.587,与理论值+1、-2有很大偏差,也是因为与配体及金属之间发生大量电子转移的原因。
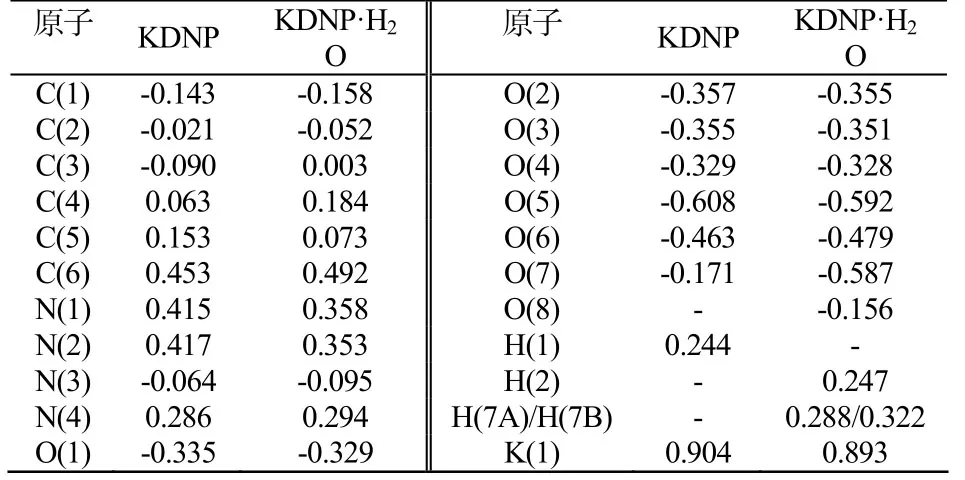
表3 B3LYP/DNP水平下原子上的净电荷Tab.3 Charge on atoms at B3LYP / DNP level
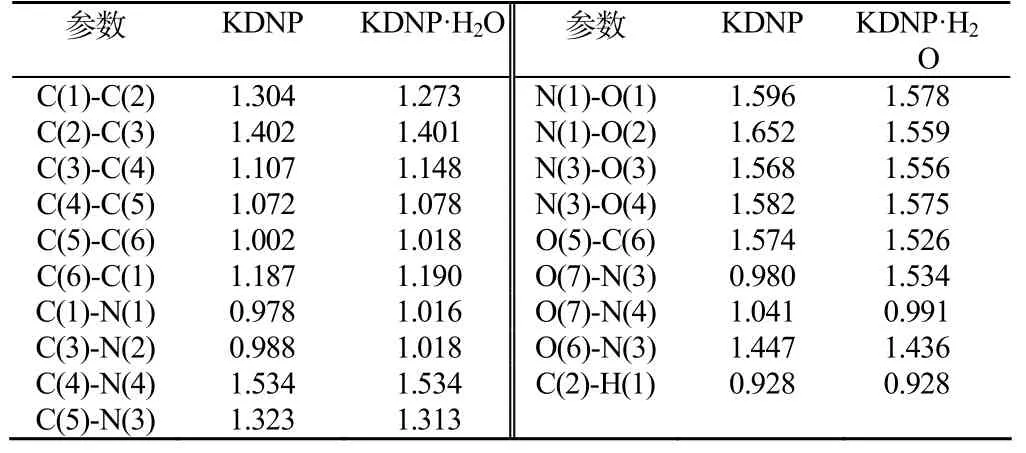
表4 B3LYP/DNP水平下KDNP与KDNP·H2O的键级Tab.4 KDNP and KDNP·H2O bond level at B3LYP / DNP level
表4 为B3LYP/DNP水平下的键级。由表4可见:KDNP与KDNP·H2O键级值分别在0.928~1.652和0.928~1.578之间,明显处于标准的单键(1.0)和标准的双键(2.0)的键级之间,推断出两种结构的共轭性较强。
2.3 静电势
图2为以分子所在平面为等值面所做的静电势等值面图。在正值区,单位正电荷一方面对化学体系的电子有吸引力,会引起电子能级下降,另一方面却有强大的核排斥力,而后者占上风,总体能量上升。从分子结构来看,点正电荷在靠近带正电荷的原子区域(C2C3C4,N7N8N11,H18,K19),静电势形成正值,此处具有亲电性;在负值区,单位正电荷一方面对化学体系的电子有强的吸引力,另一方面也有核排斥力,但前者占上风。从分子结构来看,点正电荷在靠近带负电荷原子区域(C1C5C6,N9, O10O12O13 O141O15O16),静电势形成负值,此处表现为亲核性。
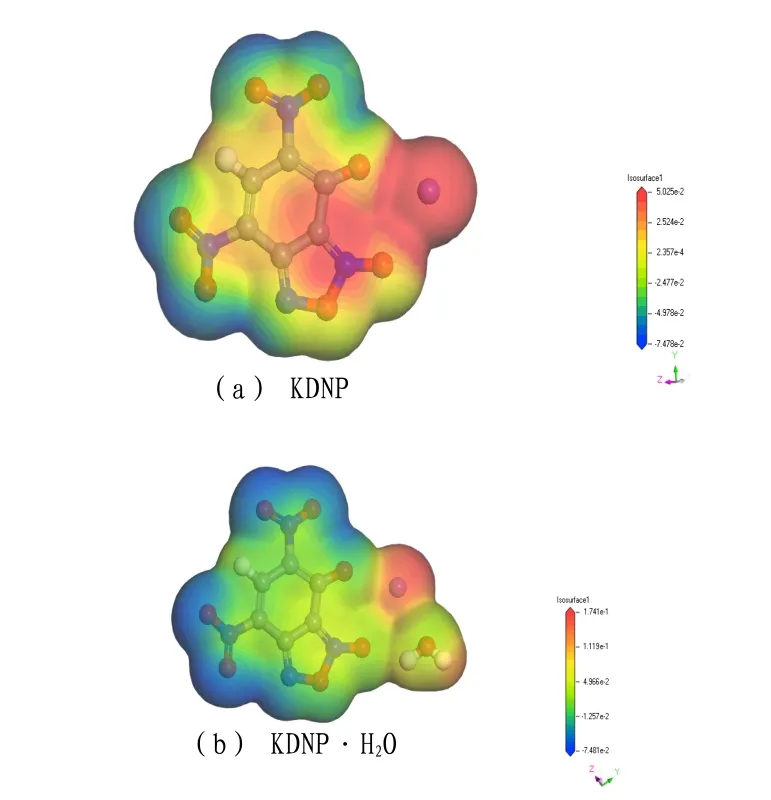
图2 静电势分布等值面Fig.2 Equivalent surface of electrostatic potential distribution
根据M. Klapötke提出的理论,化合物分子所呈现的正和负的静电势的相对大小及其伸展程度是影响其撞击感度的决定性因素[2]。从图2中可以看出KDNP·H2O较KDNP的电子密度分布均匀性较低,分子结构中心的电子缺陷较明显(即更高的正静电势),预测出其具有较高的撞击感度。
2.4 前线轨道能量分析
根据KDNP分子与轨道能级计算可知,KDNP的最高占据轨道为140号,最低空轨道为141号。由计算所得到的前线轨道能量(EHOMO,ELUMO)和能差ΔE 分别为-6.675eV,-3.271eV和3.404eV。HOMO轨道和LUMO轨道如图3所示。
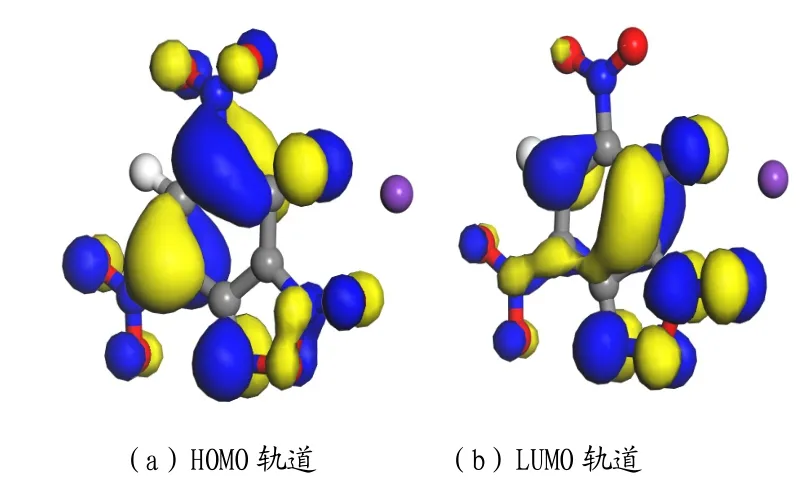
图3 KDNP 分子的HOMO轨道和LUMO轨道分布图Fig.3 HOMO orbit and LUMO orbit distribution of KDNP
根据KDNP·H2O分子与轨道能级计算可知,KDNP·H2O的最高占据轨道为150号,最低空轨道为151号。由计算所得到的前线轨道能量(EHOMO,ELUMO)和能差ΔE 分别为-6.653eV,-3.278eV和3.375eV。HOMO轨道和LUMO轨道如图4所示。
图4 KDNP·H2O分子的HOMO轨道和LUMO轨道分布图Fig.4 HOMO orbit and LUMO orbit distribution of KDNP·H2O
2.5 两种分子感度的预测
最小跃迁法研究的是炸药的最高占有轨道能量EHOMO与最低未占有空轨道能量ELUMO之差的绝对值ΔE与感度的关系,ΔE越小,则电子越容易从最高占有轨道HOMO向最低未占有空轨道LUMO跃迁,即能隙越小,电子越易跃迁,化合物稳定性越小,感度越大。研究表明对于结构相似的一类炸药,ΔE与感度之间存在着相同的变化趋势,且与键级存在相同的变化趋势[3]。表5为KDNP与KDNP·H2O分子的能隙值。据表5可知,KDNP·H2O的理论撞击感度高于KDNP。
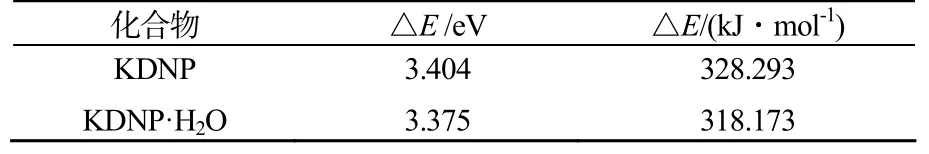
表5 两种分子的能隙值Tab.5 Energy gap values of the two molecules
2.6 两种晶体爆轰性质的计算
查阅CCDC数据库,得到KDNP的晶体构型为单斜晶系,空间点群P21/C,晶胞参数:a=7.478 9Å,b=9.899 9Å,c=12.839Å,α=90°,β=98.945°,γ=90°,构建KDNP的晶体结构模型如图5所示。根据晶体衍射实验,得到KDNP·H2O的晶体构型为三斜晶系,空间点群P-1晶胞参数a=4.567(1) Å,b=10.617(2)Å,c=11.769(3)Å,α=115.545(3)°,β=95.629(3)°,γ=91.429(3)°,晶体结构模型如图6所示。
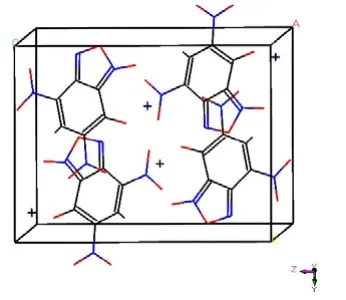
图5 KDNP的晶体结构Fig.5 Crystal structure of KDNP
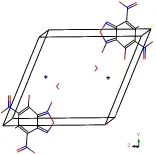
图6 KDNP·H2O的晶体结构Fig.6 Crystal structure of KDNP·H2O
2.6.1 爆炸参数的理论计算
目前最新的计算方法是计算化合物的总能量与推测爆炸分解产物的总能量之差,来推导爆炸反应所放出来的热焓值[4]。
计算化合物的总能量的量化计算采用了Dmol3模块,选用了GGA/PBE泛函,K点取样的间隔是0.05 A-1,进行结构优化计算,求得反应物与分解产物的能量差△Edet。接着用修正公式△Hdet=1.127*△Edet+ 0.046,求得反应物的分解焓值△Hdet。
爆炸产物的预测根据是产物能量最低、最稳态的原理。其中,活泼金属形成金属氧化物;氮生成氮气;氢则首先和氧生成水,剩下的氧与碳形成二氧化碳;若氧不足,多余的碳以固态存在。对于KDNP,1个晶胞内的爆炸反应方程式为:
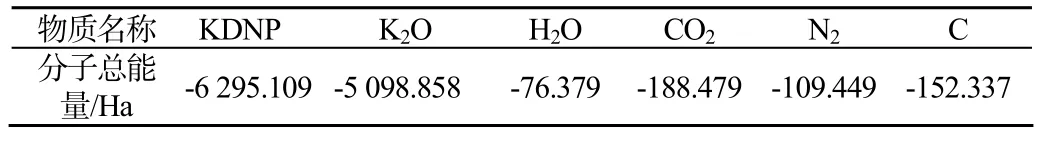
表6 所需物质的分子总能量Tab.6 Total energy required two moculer
Etot=(-5 098.858÷4×2)+(-76.379×2)+(-188.479 ×12)+(-109.449×8)+(-152.337÷4×12)-(-6 295.109)=-1.431Ha=-898.143 kcal/mol =-0.801 407 36 kcal/g
代入修正公式:Y=1.127x+0.046
得爆热为0.949 kcal/g,既3.973 kJ/g。
对于KDNP·H2O,1个晶胞内爆炸反应方程式为:2C6H3N4O8K→K2O(S)+3H2O+6CO2+4N2+6C(S)
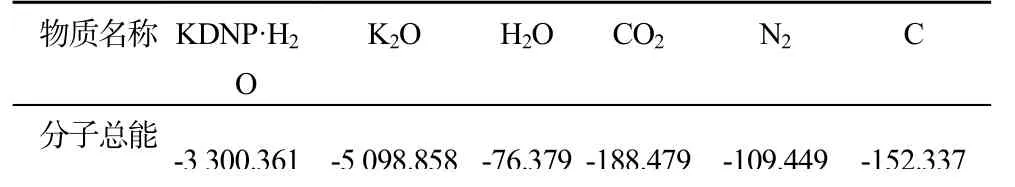
表7 所需物质的分子总能量Tab.7 Total energy required two moculer
Etot=(-509 8.858÷4×2)+(-76.379×3)+(-188.479×6)+(-109.449×4)+(-152.338÷4×6)-(-3 300.361)=-0.666Ha=-418.032kcal/mol =-0.701kcal/g
代入修正公式:Y=1.127x+0.046得爆热为0.836 kcal/g,既3.500 kJ/g。
2.6.2 爆速爆压的理论计算
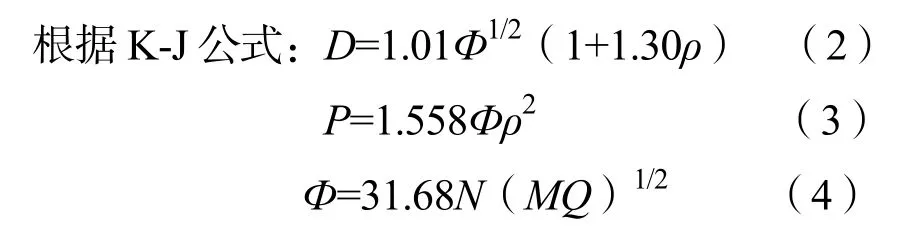
式(2)~(4)中:N为每克爆炸物释放的气体摩尔数,mol/g;M为释放的气体的平均分子量,g/mol;Q为爆热,kcal/g;ρ为密度,g/cm3;D为爆速,km/s;P为爆压,GPa 。具体数值如表8所示。

表8 KDNP及KDNP·H2O的爆轰性质Tab.8 Detonation properties of KDNP and KDNP·H2O
3 结论
通过对7-羟基-4,6-二硝基-5-氢-苯并呋咱钾(KDNP)和7-羟基-4,6-二硝基-5-氢-苯并呋咱钾的一水化物(KDNP·H2O)的量子化学计算,得到几何构型参数、电荷分布、键级、静电势和前线轨道分布,并进行了分析。几何构型分析表明:KDNP和KDNP·H2O分子共面性较好,结构较稳定,具有一定的共轭性,是一种离子型化合物。静电势表明了KDNP·H2O分子可能比KDNP分子有较高的撞击感度。前线轨道分布表明了KDNP·H2O分子的能差比KDNP小;根据以上的计算结果,利用最易跃迁法对两种分子的感度进行了预测,表明KDNP·H2O较KDNP敏感;KDNP晶体和KDNP·H2O晶体的爆热值分别为3.973kJ/g、3.500kJ/g,K-J公式计算得到爆压分别为22.223GPa、21.252GPa,爆速分别为6.884km/s、6.774km/s,表明KDNP·H2O晶体的爆轰性能与KDNP晶体基本在同一水平。
[1]Fronabarger J W, Williams M D, Sanborn W B, et al. KDNP–A lead free replacement for lead styphnate[J]. Propellants, Explosives, Pyrotechnics, 2011, 36(5): 459-470.
[2]Thomas M. Klapötke.Chemistry of high-energy materials[M]. Walter de Gruyter GmbH & Co KG,2015.
[3]张朝阳, 舒远杰, 赵晓东,等. 分子内氨基对C-NO2影响的理论研究[J]. 火炸药学报, 2004, 27(3):32-35.
[4]Bushuyev O S, Brown P, Maiti A, et al. Ionic polymers as a new structural motif for high-energy-density materials[J]. Journal of the American Chemical Society,2012,134(3): 1 422-1 425.
The Properties Prediction of KDNP and KDNP·H2O by Using Quantum Chemistry Calculation
ZHANG Shi-xin, SHENG Di-lun, ZHU Ya-hong, NI De-bin , SHI Sheng-nan, PANG Cong-cong
(Shaanxi Applied Physics and Chemistry Research Institute, Xi’an, 710061)
The molecular properties prediction of KDNP and KDNP·H2O based on Materials Studio software by Dmol3 module with the process of B3LYP/DNP was carried out, and the energies of detonation of KDNP and KDNP·H2O were calculated respectively. It showed that KDNP molecular structures are coplanar, but the H2O molecule in KDNP·H2O is different from the furoxan ring with a dihedral angle of 29.35°. Computed band order suggests that the whole molecular are strongly conjugate. Electrostatic potential infers that the impact sensitivity of KDNP· H2O may be higher than that of KDNP. The calculated frontier orbital energy differences of KDNP·H2O is smaller than that of KDNP, so it is believed that KDNP·H2O is more sensitive than KDNP. Meanwhile, the simulated detonation properties of KDNP·H2O is similar to KDNP.
KDNP;KDNP·H2O;Quantum chemistry;Sensitivity;Detonation properties
TQ563
A
1003-1480(2016)06-0048-05
2016-08-29
张诗欣(1992 -),女,在读硕士研究生,主要从事新型火工药剂的合成与应用研究。
