Miller-Dieker综合征产前诊断1例
2016-02-13郭莉卢建陈汉彪王挺郑来萍任丛勉尹爱华
郭莉 卢建 陈汉彪 王挺 郑来萍 任丛勉 尹爱华
(广东省妇幼保健院 医学遗传中心,广东 广州 511442)
Miller-Dieker综合征产前诊断1例
郭莉 卢建 陈汉彪 王挺 郑来萍 任丛勉 尹爱华
(广东省妇幼保健院 医学遗传中心,广东 广州 511442)
Miller-Dieker综合征是一种少见的常染色体遗传病,临床上以中枢神经系统异常,包括无脑回、小头畸形、胼胝体缺如或发育不良为特点,伴有特征性面容等先天性异常的临床综合表现[1]。Miller-Dieker综合征属于无脑回畸形(agyria)中一类。作为一种严重的大脑皮层发育畸形疾病,其发病率大约为1/13 000 ~20 000,患儿预后多数较差[2],产前发现并通过介入性诊断病例少有报道。本文结合广东省妇幼保健医院医学遗传中心诊治的1 例孕妇产前诊断无脑回畸形胎儿,并对相关文献进行复习,探讨Miller-Dieker综合征的临床特点及致病基因,以提高对产前胎儿无脑回畸形的认识。
1 病例摘要
孕妇28岁,G5P0A4,因“停经23周,超声检查发现胎儿双侧脑室增宽”就诊。夫妇双方既往体健,非近亲,否认家族史,否认孕期有害毒物接触和传染病史。 孕妇初婚,此为第5次妊娠,曾胎死宫内2次,分别为孕3月和孕5月,宫外孕1次,自然流产1次。以往流产胎儿均未进一步检查。此孕期规律产检,孕23周超声检查发现胎儿左右侧脑室分别为13.3mm和10.9mm,头颅MRI发现胎儿无脑回、大脑皮层发育异常、双外侧裂形成不良、双侧脑室扩大。行羊膜腔穿刺,羊水染色体G显带和SNP检测,常规核型结果提示46,XX (图1),SNP发现17号染色体17p13.3位置存在2.78Mb片段缺失(图2)。夫妇双方外周血染色体未见异常。由于胎儿预后不良,夫妇要求回当地医院终止妊娠,未予尸检。

图1 羊水染色体G显带核型结果
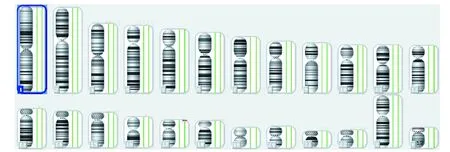
图2 羊水SNP检测17号染色体17p13.3位置存在2.78Mb片段缺失
2 讨论
Miller-Dieker综合征是属于Ⅰ型经典型无脑回畸形,由位于17号染色体上17p13.3位置LIS1基因(PAFAH1B1; OMIM 601545)缺失或突变引起[3]。Miller-Dieker综合征主要表现为无脑回、小头畸形、胼胝体缺如或发育不良、特征性面容以前额突出、短鼻、上翘鼻孔、外凸的上唇为特点。由于大脑发育不全,会导致严重智力低下、早期肌张力增高、角弓反张、痉挛、癫痫发作等[4]。
产前Miller-Dieker综合征病例主要以无脑回、皮质发育延迟等脑部发育异常为特点,由Saltzman[5]于1991年首次报道。该报道包括两病例,其中一例孕25周发现羊水过多,孕31周超声发现胎儿无脑回,伴有无脑回家族史,且孕妇携带平衡易位染色体17p13;另一例孕31周发现胎儿宫内生长受限,伴有严重羊水过多,法洛四联征,无脑回等超声表现。Okamura[6]报道2例产前诊断无脑回畸形(胎儿染色体正常),胎儿MRI显示大脑表面光滑,但中期超声未能发现,随后该两例分别于孕24周和31周胎儿MRI检查发现双侧脑室扩大,皮层的表面光滑。Fong等[7]通过对3例产前超声检查怀疑无脑回畸形的前瞻性研究,分别在孕23、26和30周进行超声检查,随后通过产前MRI证实无脑回。
从解剖学研究显示,有些大脑半球内侧面的,脑沟早在孕16~19周就可以辨认,但有研究[8]发现,有经验的超声医生可以比早期超声医学文献所认为的孕27~28周要更早发现大脑半球内侧面的脑沟,因此常规孕期18~22周行超声检查很难发现无脑回畸形,因为孕24周前大脑表面光滑仍可能为正常表现,因此在孕24周前产前诊断无脑回畸形非常困难[9],因而为了早期诊断,需要关注其他相关的症状以助诊断,如产前超声检查可发现羊水过多、宫内生长受限、轻度脑室扩张,胼胝体发育不全、小头畸形,而罕见包括小颌畸形,先天性心脏病、泌尿生殖系统畸形,脐膨出等[7,10]。Chen[11]报道羊水过多、胎儿宫内生长受限和侧脑室增宽是最常见的相关的超声异常,分别占66%,62%和59%;无脑回畸形、胼胝体发育不全或缺如分别占41%和17%,且分别都在孕晚期发现;圆锥动脉干畸形占14%,脐膨出和肾功能异常占7%。但如果发现其他头颅超声异常如侧脑室增宽、胼胝体发育不全等,需行胎儿头颅MRI检查,以协助诊断。本病例孕23周超声发现胎儿双侧脑室增宽,胎儿头颅MRI发现胎儿无脑回、大脑皮层发育异常、双外侧裂形成不良,均为报道常见的临床表现之一。
由于Miller-Dieker综合征与17p13.3片段缺失相关,由17号染色体发生易位或倒位所致,可通过父母平衡易位携带而遗传,或新发突变引起。曾有研究估计,约80%的病例是新发异常,约20%遗传自父母重排染色体[12]。Chen[11]研究29例Miller-Dieker综合征病例中,产前诊断羊水染色体核型未见异常占24%,介入指征包括高龄、习惯性流产、超声异常,意味着仅使用常规核型分析,容易导致该疾病漏诊。其研究提示至少45%病例与父母染色体隐匿易位或倒位相关。
本研究病例孕妇产前超声检查和胎儿头颅MRI提示胎儿侧脑室增宽、无脑回、大脑皮层发育异常、双外侧裂形成不良,羊水SNP检测提示17p13.3位置发生 2.78M片段缺失,包含已知基因54个,符合Miller-Dieker 综合征诊断。羊水染色体核型结果提示46,XX未发现异常,考虑常规染色体检查显微镜下能分辨片段大小为5~10Mb,由于该病例缺失片段大小2.78Mb,因此缺失片段肉眼镜下不能分辨。由于该对夫妇既往胎死宫内2次,夫妇双方可能存在染色体微小片段的平衡易位。因此,该对夫妇下次妊娠仍建议产前胎儿SNP检测。
由于Miller-Dieker综合征胎儿出生后预后差,因此产前诊断该疾病显得非常重要。产前检查发现胎儿无脑回、胼胝体缺如或发育不良、巨脑回、小头畸形、大脑皮质发育异常、双外侧裂形成不良,是Miller-Dieker综合征重要的产前影像学指标,应该考虑可能存在17号染色体17p13.3缺失。如产前超声检查发现胎儿侧脑室增宽,羊水过多,胎儿宫内发育迟缓,需监测脑部发育,并进一步行包括脑沟和裂缝的详细检查,和介入性产前诊断。胎儿染色体核型正常时,应进一步行CMA检测,以便及早确诊,以排除Miller-Dieker综合征,明确诊断后建议终止妊娠。胎儿头颅MRI和CMA检测对产前诊断胎儿Miller-Dieker综合征具有积极作用。
[1] 陆国辉.产前遗传病诊断[M].广州:广东科技出版社.2002.
[2] Dobyns WB, Reiner O, Carrozzo R, et al. Lissencephaly. A human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13[J]. JAMA, 1993, 270(23): 2838-2842.
[3] Reiner O, Carrozzo R, Shen Y, et al. Isolation of a Miller-Dieker lissencephaly gene containing G protein β-subunitlike repeats[J]. Nature,1993,364:717-721.
[4] Jones KL. Miller-Dieker syndrome[M]. Philadelphia: Elsevier Saunders.2006.
[5] Saltzman DH, Krauss CM, Goldman J M, et al. Prenatal diagnosis of lissencephaly[J]. Prenat Diagn 1991;11: 139-143.
[6] Okamura K, Murotsuki J, Sakai T, et al. Prenatal diagnosis of lissencephaly by magnetic resonance image[J]. Fetal Diagn Ther,1993,8:56-59.
[7] Fong KW, Ghai S, Toi A, et al. Prenatal ultrasound findings of lissencephaly associated with Miller-Dieker syndrome and comparison with pre- and postnatal magnetic resonance imaging[J]. Ultrasound Obstet Gynecol,2004,24:716-723.
[8] 陈晓康,林惠通,吕国荣.超声观察胎儿大脑沟回发育及其临床意义[J].中华超声影像学杂志,2009,18(2):149-152.
[9] 巫敏,王慧芳.胎儿无脑回-巨脑回畸形产前诊断的研究进展[J/CD].中国产前诊断杂志(电子版),2016,8(1):36-41.
[10] Ghai S, Fong KW, Toi A, et al. Prenatal US and MR imaging findings of lissencephaly: review of fetal cerebral sulcal development[J]. Radiographics,2006,26:389-405.
[11] Chen CP,Chang TY,Guo WY,et al.Chromosome 17p13.3 deletion syndrome: aCGH characterization,prenatal findings and diagnosis, and literature review[J].Gene,2013,532(1):152-159.
[12] Dobyns WB, Das S. GeneReviews[M]. Seattle (WA) :University of Washington.2014.
编辑:宋文颖
10.13470/j.cnki.cjpd.2016.04.020
R714.55
B
2016-10-15)
