一种基于苯并噻二唑衍生物的F-荧光探针分子的理论研究
2016-01-31李云飞王新收吴文鹏
李云飞, 王新收,吴文鹏
(1. 河南医学高等专科学校 药学系, 河南 郑州 451191; 2. 河南大学 民生学院, 河南 开封 475004;
3. 河南大学 化学化工学院, 环境与分析科学研究所, 河南 开封 475004)
一种基于苯并噻二唑衍生物的F-荧光探针分子的理论研究
李云飞1, 王新收2,吴文鹏3*
(1. 河南医学高等专科学校 药学系, 河南 郑州 451191;2. 河南大学 民生学院, 河南 开封 475004;
3. 河南大学 化学化工学院, 环境与分析科学研究所, 河南 开封 475004)
摘要:用密度泛函理论研究了最近实验上合成的一种基于苯并噻二唑衍生物的F-荧光探针(1)的性质. 通过对相关化学反应热力学参数的计算,提出了1对F-具有高选择性的可能原因;同时用含时密度泛函理论对电子吸收光谱和荧光发射光谱进行了理论计算,解释了实验光谱.
关键词:氟离子;荧光探针;苯并噻二唑;密度泛函理论;含时密度泛函理论
氟是电负性最大的元素,氟离子是电荷半径比最小的阴离子,具有独特的化学性质. 氟离子在龋齿的防治、骨质疏松症的临床治疗、食品科学和环境科学等领域中都起着重要作用[1-4]. 因此,对氟离子的快速检测引起了人们广泛的关注. 荧光化学传感器是以荧光信号作为宏观信号的化学传感器,它具有操作简单、灵敏度高、选择性强、检测限低、方便快捷等优点,因此得到了广泛的应用[2-3].
最近,喻艳华等[5]合成了一种荧光化学传感器,其中的荧光探针分子是基于苯并噻二唑衍生物1(图1). 在乙腈和水的混合溶液(体积比9∶1)中,1的最大吸收峰为376 nm,当加入F-后最大吸收峰的强度显著减弱,峰位置蓝移至360 nm;最大荧光发射峰强度减弱60%,峰位置从455 nm蓝移至435 nm. 当加入其他阴离子,如Cl-、Br-、CN-、NO3-、AcO-、HSO4-等时,荧光光谱没有发生明显变化. 该传感器对F-有非常好的选择性,作者推测F-能脱除1中的三甲基硅基,发生反应(1),生成2(图1),而其他离子不能脱除三甲基硅基. 为了更深入地认识这一现象,了解光谱变化的实质,本文作者从理论上对这一反应进行了研究,并模拟化合物1和2的电子吸收光谱和荧光光谱. 前期研究[6-9]表明,密度泛函理论在预测分子的结构和光谱方面表现出色. 因此,本文作者用密度泛函理论研究荧光探针1的性质.
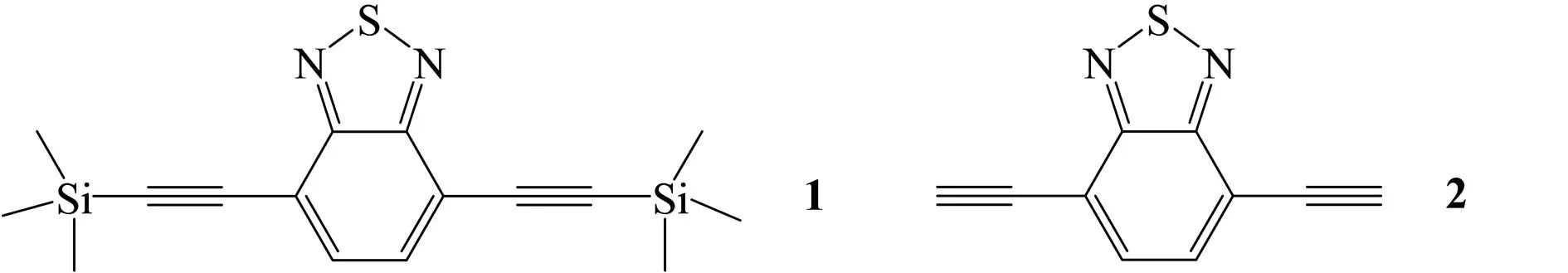

(1)
图1荧光探针1和化合物2的结构式及可能的化学反应方程式
Fig.1Structuresoffluorescentprobe1andcompound2andpossiblechemicalreactionequations
1计算方法
在B3LYP[10-11]/6-31G*[12-13]水平上优化得到基态的几何结构,在该构型的基础上得到相应的热力学参数. 化合物1和2的第一激发单重态的几何结构是在TD[14-15]-B3LYP/6-31G*水平上优化得到的,它们的垂直激发能和垂直发射能在相同水平上计算得到.所有计算都是在Gaussian09程序包[16]中完成的, 并用CPCM模型[17-18]考虑了乙腈溶剂的影响.
2结果与讨论
2.1 几何结构
从优化得到的化合物1和2的几何结构(基态和第一激发单重态)可以看出,化合物1和2的炔基苯并噻二唑部分的原子共平面. 主要的键长参数绘于图2中. 从图2中基态结构可以看出,化合物1脱去三甲基硅基变成化合物2后,C≡C键长缩短0.001 2 nm,C-C键长增加0.000 3 nm,苯环上键长变化小于0.000 2 nm,噻二唑环键长几乎不变. 由此可推断1变成2后电子离域程度减小,电子吸收光谱可能发生蓝移. 比较图2中激发态和基态结构可以看出,1激发态中C≡C键长比基态增加0.000 3 nm,C-C键长缩短0.002 7 nm,苯并噻二唑部分键长变化较大,最大的为0.005 4 nm. 化合物2激发态键长变化规律与1相似,不再一一赘述.
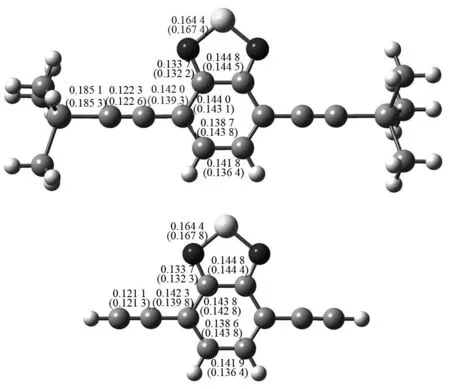
括号外为基态键长;括号内为第一激发态键长,单位nm.图2 优化得到的化合物1(上)和2(下)的主要键长Fig.2 The optimized bondlengths of compounds 1 (top) and 2 (bottom)
2.2 热力学参数
计算得到的反应式(1)中各反应的能量变化(ΔE),焓变(ΔH)和吉布斯自由能变化(ΔG)列于表1. 从表1可以看出,当X-为F-时,ΔH和ΔG都是最小的,并且为负值,其他几种离子参与的反应ΔH和ΔG都较大. 从热力学上分析,F-更易发生反应,其他几种离子不太容易发生反应. 这可能就是荧光探针1对F-有较高选择性的原因之一.
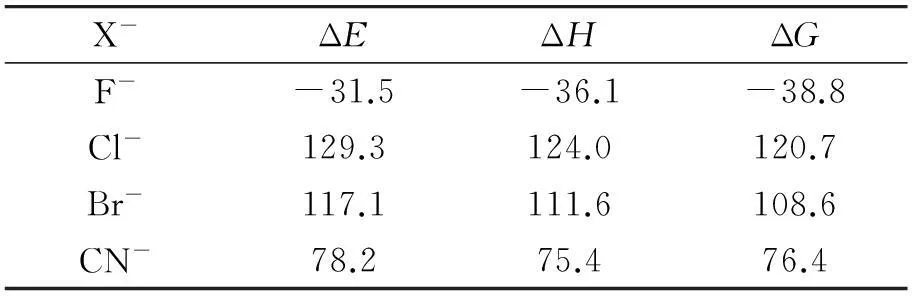
表1 反应(1)的热力学参数(kcal/mol)
2.3 电子吸收光谱
众所周知,电子吸收光谱的性质和前线分子轨道密切相关,我们将化合物1和2的最高占据轨道(HOMO)和最低空轨道(LUMO)轮廓图绘于图3中. 从图3可以看出,HOMO主要分布在炔基、苯环和氮原子上,LUMO在硫原子上也有分布. 从整体上看,1和2的HOMO和LUMO形状分别相似. 计算得到的电子吸收光谱数据列于表2中. 显然,无论是峰的位置(λ)还是相对强度(振子强度f),理论计算值和实验值[5]符合得都很好. 从表2还可以看出,第一激发态主要来自于HOMO→LUMO的跃迁. 1和2的LUMO能级虽相近,但由于1脱去三甲基硅基变成2后HOMO能级降低,引起HOMO-LUMO能级差ΔEHL增大,从而使第一激发态的垂直激发能(ΔEa)变大,光谱蓝移了20 nm. 这一结果与前文中根据几何结构的预测相一致,同时这也与实验光谱蓝移值16 nm[5]很接近,验证了实验预测的合理性(即1反应后生成了2).

图3 化合物1和2的前线分子轨道轮廓图Fig.3 The frontier molecular orbitals of compounds 1 and 2

CompoundTransitionConfigurationcoefficientHOMO/eVLUMO/eVΔEHL/eVΔEa/eVλa/nmfλExpt./nm[5]1HOMO→LUMO0.701-6.09-2.833.263.273800.5933762HOMO→LUMO0.702-6.26-2.843.423.443600.345360
2.4 荧光光谱
计算得到的荧光光谱数据列于表3. 从表3可以看出,荧光光谱主要来自于LUMO→HOMO的跃迁. 荧光光谱与吸收光谱相似,2相对于1,光谱发生蓝移,荧光强度明显减弱. 这与实验光谱[5]一致.

表3 化合物1和2的荧光光谱数据
3结论
用密度泛函理论和含时密度泛函理论优化得到了荧光探针分子1基态和第一激发态的几何结构,并得到了相关化学反应的热力学参数,同时也从理论上预测了1的电子吸收光谱和荧光发射光谱. 结果表明,从基态到激发态,1的单键缩短,叁键伸长,共轭程度增强. 从热力学上分析,1与F-反应,脱去三甲基硅基生成2,该反应的ΔH和ΔG对反应发生都是有利的,这可能是1对F-有高选择性的原因之一. 从电子吸收光谱和荧光发射光谱看,1变成2后,光谱都发生了蓝移,与实验观测一致. 光谱蓝移的原因是1脱去三甲基硅基后HOMO能级降低,LUMO能级基本不变,从而使HOMO-LUMO能隙增大.
参考文献:
[1] 郝一莼,孙小单,徐桐,等. 氟离子检测分析方法研究进展[J]. 中国地方病防治杂志,2010,25(6): 412-415.
[2] 吴振,周应,任君. 氟离子荧光化学传感器的研究进展[J]. 化学传感器,2012,32(1): 41-52.
[3] 鲍寅寅,白如科. 基于有机硅化合物的反应型氟离子荧光化学传感器[J]. 化学进展,2013,25(2/3): 288-295.
[4] RAO M R, MOBIN S M, RAVIKANTH M. Boron-dipyrromethene based specific chemodosimeter for fluoride ion [J]. Tetrahedron, 2010, 66: 1728-1734.
[5] 喻艳华,付成. 用基于苯并噻二唑衍生物的比率荧光化学传感器检测氟离子[J]. 化学研究, 2014, 25(5): 482-487.
[6] 宁攀, 赵建想. 利用密度泛函理论研究α-联噻吩体系H(C4H2S)nH的结构和电子光谱[J]. 化学研究, 2013, 24(5): 493-500.
[7] 李洁琼, 赵清岚. 三种席夫碱-Ni(Ⅱ)配合物的电子结构和吸收光谱的理论计算[J]. 化学研究, 2014, 25(5): 497-503.
[8] 吴文鹏, 曹艳. 三(4-硝基苯基)甲烷衍生物的结构和振动光谱的理论研究[J]. 化学研究, 2014, 25(6): 609-615.
[9] 李洁琼, 李永红. 用密度泛函理论研究两种金属镍席夫碱配合物的电子结构和光谱性质[J]. 化学研究, 2014, 25(6): 616-621.
[10] BECKE A D. Density-functional thermochemistry. III. The role of exact exchange [J]. J Chem Phys, 1993, 98(7): 5648-5652.
[11] LEE C, YANG W, PARR R G. Development of the Colic-Salvetti correlation-energy figure into a functional of the electron density [J]. Phys Rev B, 1988, 37: 785-789.
[12] PETERSSON G A, BENNETT A, TENSFELDT T G, et al. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row atoms [J]. J Chem Phys, 1988, 89: 2193-2218.
[13] PETERSSON G A, AL-LAHAM M A. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms [J]. J Chem Phys, 1991, 94: 6081-6090.
[14] FURCHE F, AHLRICHS R. Adiabatic time-dependent density functional methods for excited state properties [J]. J Chem Phys, 2002, 117: 7433-7447.
[15] SCALMANI G, FRISCH M J, MENNUCCI B, et al. Geometries and properties of excited states in the gas phase and in solution: Theory and application of a time-dependent density functional theory polarizable continuum model [J]. J Chem Phys, 2006, 124: 094107 1-15.
[16] FRISH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09 [CP]. Revision A.02, Wallingford CT: Gaussian, Inc., 2009.
[17] BARONE V, COSSI M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model [J]. J Phys Chem A, 1998, 102: 1995-2001.
[18] COSSI M, REGA N, SCALMANI G, et al. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model [J]. J Comp Chem, 2003, 24: 669-681.
[责任编辑:任铁钢]
Theoretical investigations on a F-fluorescent probe based on
benzotiadiazole derivative
LI Yunfei1, WANG Xinshou2, WU Wenpeng3*
(1.DepartmentofPharmacology,HenanMedicalCollege,Zhengzhou451191,Henan,China;2.MinshengCollege,
HenanUniversity,Kaifeng475004,Henan,China;3.InstituteofEnvironmentalandAnalyticalSciences,
CollegeofChemistryandChemicalEngineering,HenanUniversity,Kaifeng475004,Henan,China)
Abstract:The properties of a F-fluorescent probe synthesized recently based on benzotiadiazole derivative (1) were investigated by use of density functional theory. According to the calculated related chemical reaction thermodynamic parameters, the possible reason for the high selectivity of 1 on F-was proposed. Furthermore, the electronic absorption spectra and fluorescent emission spectra were predicted by time dependent density functional theory, which was used to explain the experimental spectra.
Keywords:F-; fluorescent probe; benzotiadiazole; density functional theory; time dependent density functional theory
作者简介:李云飞(1982-), 男, 讲师, 研究方向为光谱分析.*通讯联系人, E-mail:wenpengwu@126.com.
基金项目:河南大学博士科研启动基金(B2013141).
收稿日期:2014-06-04.
文章编号:1008-1011(2015)06-0575-04
中图分类号:O641
文献标志码:A
