5-取代-2,3-二氢-1H-[1,4]二氮艹卓类化合物的合成*
2016-01-17陈启凡张慧东武晓燕辽东学院化学工程学院辽宁丹东8003东北师范大学化学系吉林长春0004
陈启凡,张慧东,武晓燕,2,刘 飞(.辽东学院化学工程学院,辽宁丹东 8003; 2.东北师范大学化学系,吉林长春 0004)
5-取代-2,3-二氢-1H-[1,4]二氮艹卓类化合物的合成*
陈启凡1,张慧东1,武晓燕1,2,刘飞1
(1.辽东学院化学工程学院,辽宁丹东118003; 2.东北师范大学化学系,吉林长春110004)
摘要:以乙醇为溶剂,烯胺酮(1)和乙二胺(2)经Michael亲核加成反应合成了7个5-取代-2,3-二氢-1H-[1,4]二氮艹卓类化合物(3a~3g),其中3a~3d为新化合物,其结构经1H NMR,IR和元素分析表征。在最佳反应条件[1 30 mmol,n(2)∶n(1)=1.2,回流反应8 h~10 h,无水乙醇为重结晶溶剂]下合成的3a~3g,收率70.0%~74.2%。
关键词:烯胺酮; Michael亲核加成反应;二氮艹卓;合成
杂环化合物是医药、农药、染料和精细化工品的重要中间体。其中,含氮的杂环化合物具有潜在的生物活性。二氮杂艹卓化合物具有二氮杂七元环结构,显示出重要的药理学活性,自19世纪被发现以来,引起科学家的极大关注[1-4]。已报道的二氮杂艹卓化合物主要有咪唑并二氮杂艹卓、苯并二氮杂艹卓、嘧啶并二氮杂艹卓、吡唑并二氮杂艹卓、噻吩并二氮杂艹卓等等[5-7]。其中,苯并二氮杂艹卓类化合物具有极其广泛的药理活性,是最先被确定为特权结构的一类化合物。此类化合物具有极高的研究价值和广阔的应用前景,是临床上应用较多的安眠药,但在临床长期应用中,也渐显露出制约其在使用上的征象[8]。为了减少不良反应,人们开发了一类新的催眠药,如非苯二氮艹卓类化合物,由于起效快,药效强,副作用小,应用越来越广泛,取得了可喜的疗效[9]。
为目前为止,合成非苯二氮艹卓类单环化合物的文献方法较少,仅见Hassanien等[10]以烯胺酮为原料合成了1,2,3-三氢-[1,4]二氮艹卓化合物。本文在文献[10]方法的基础上,根据Michael亲核加成反应原理[11],以烯胺酮(1a~1g)和乙二胺(2)为原料,经Michael亲核加成反应,合成了7 个5-取代-2,3-二氢-1H-[1,4]二氮艹卓类化合物(3a~3g),其中3a~3d为新化合物,其结构经1H NMR,IR和元素分析表征。并考察了原料配比,反应体系的酸碱性及重结晶溶剂对反应的影响。在最佳反应条件[1 30 mmol,n(2)∶n(1)=1.2,回流反应8 h~10 h,无水乙醇为重结晶溶剂]下合成的3a~3g,收率70.0%~74.2%。
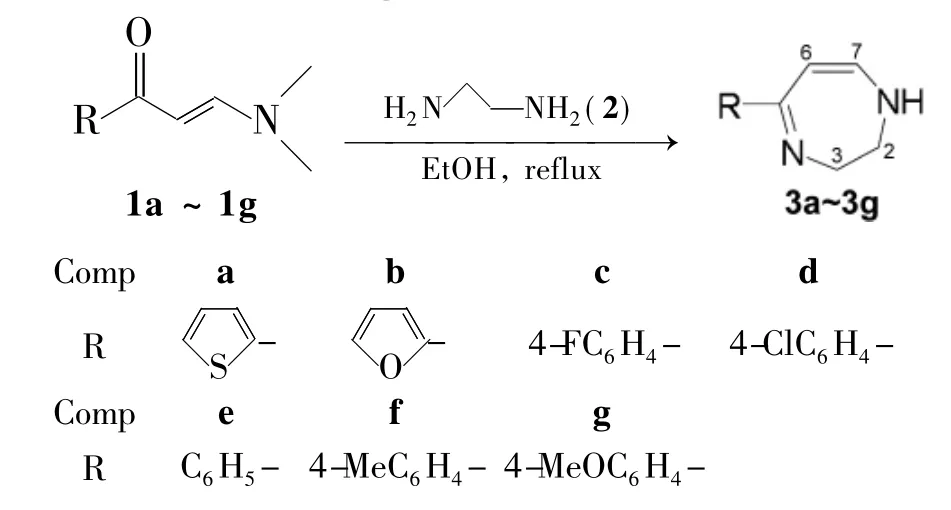
Scheme 1
1 实验部分
1.1仪器与试剂
X-5型熔点仪(温度未校正); Avance Π 400 HMz型核磁共振仪(CDCl3为溶剂,TMS为内标); Spectrum 100型红外光谱仪(KBr压片); Perkin Elmer 2400型元素分析仪。
所用试剂均为分析纯,国药集团化学试剂有限公司;其中2使用前经蒸馏除水处理。
1.2 3a~3g的合成(以3a为例)
在反应瓶中加入3-(N,N-二甲胺基)-1-(2-噻吩基)-2-丙烯-1-酮(1a)5.46 g(30 mmol)和无水乙醇10 mL,搅拌使其溶解;缓慢将2 2.1 mL(36 mmol)的无水乙醇(5 mL)溶液滴入反应液底部,滴毕,回流反应约2 h时出现白色沉淀,再反应8 h(TLC检测)。冷却至室温,静置,抽滤,滤饼用蒸馏水洗涤2~3次,真空干燥(60℃/80 kPa),用无水乙醇重结晶得3a。
用类似的方法合成3b~3g。
5-(2-噻吩基)-2,3-二氢-1H[1,4]二氮艹卓(3a):淡黄色固体,收率73%,m.p.194.6℃~194.8℃;1H NMR δ:3.41(t,J=3.5 Hz,4H,2,3-H),5.60(d,J=7.5 Hz,1H,6-H),6.81(q,J=7.5 Hz,1H,7-H),7.07(t,J=4.5 Hz,1H,Thi-H),7.48(d,J=5.0 Hz,1H,Thi-H),7.54(d,J=3.5 Hz,1H,Thi-H),10.04(t,J=5.0 Hz,1H,NH); IR ν:3 088,1 645,1 559,1 510,1 273,1 220,852,774 cm-1; Anal.calcd for C9H10N2S:C 60.64,H 5.65,N 15.72; found C 60.61,H 5.60,N 15.74。
5-(2-呋喃基)-2,3-二氢-1H[1,4]二氮艹卓(3b):褐色色固体,收率70.0%,m.p.189.2℃~191.0℃;1H NMR δ:3.42(t,J=3.0 Hz,4H,2,3-H),5.63(d,J=7.5 Hz,1H,6-H),6.47(t,J=1.5 Hz,1H,Fur-H),6.84(q,J=7.5 Hz,1H,7-H),7.00(d,J=3.5 Hz,1H,Fur-H),7.49(br,1H,Fur-H),10.09(t,J=5.0 Hz,1H,NH); IR ν:3 256,3 078,1 655,1 529,1 510,1 206,839,790,701 cm-1; Anal.calcd for C9H10N2O:C 66.65,H 6.21,N 17.27; found C 66.62,H 6.19,N 17.30。
5-(4-氟苯基)-2,3-二氢-1H[1,4]二氮艹卓(3c):淡黄色固体,收率74%,m.p.176.2℃~176.9℃;1H NMR δ:3.44(t,J=3.0 Hz,4H,2,3-H),5.67(d,J=7.5 Hz,1H,6-H),6.87(q,J=7.5 Hz,1H,7-H),7.07(t,J=9.0 Hz,2H,ArH),7.86(q,J=6.0 Hz,2H,ArH),10.32(t,J=5.5 Hz,1H,NH); IR ν:3 246,3 078,2 932,1 633,1 554,1 509,1 271,1 219,1 154,850,786 cm-1; Anal.calcd for C11H11N2F:C 77.16,H 6.48,N 16.36; found C 77.13,H 6.45,N 16.40。
5-(4-氯苯基)-2,3-二氢-1H[1,4]二氮艹卓(3d):白色固体,收率71%,m.p.217.8℃~218.4℃;1H NMR δ:3.46(t,J=3.0 Hz,4H,2,3-H),5.58(d,J=7.5 Hz,1H,6-H),6.89(q,J=7.5 Hz,1H,7-H),7.38(d,J=8.5 Hz,2H,ArH),7.79(d,J=8.5 Hz,2H,ArH),10.36(t,J=5.0 Hz,1H,NH); IR ν:3 257,3 041,2 925,1 630,1 578,1 547,1 273,1 213,1 056,864 cm-1; Anal.calcd for C11H11N2Cl:C 70.40,H 5.91,N 14.93; found C 70.38,H 5.88,N 14.95。
5-苯基-2,3-二氢-1H[1,4]二氮艹卓(3e),5-(对甲苯基)-2,3-二氢-1H[1,4]二氮艹卓(3f)和5-(4-甲氧基-苯基)-2,3-二氢-1H[1,4]二氮艹卓(3g)为已知化合物,其表征数据与文献[10]值吻合。
2 结果与讨论
为提高产物的收率,考察了诸因素对反应的影响,寻找最佳反应条件。
2.1反应体系的酸碱性
1 30 mmol,其余反应条件同1.2,考察酸(冰醋酸、盐酸)性反应体系和中性反应体系对产物收率的影响。实验结果表明:在适量冰醋酸中反应数小时,未见溶液中有晶体析出,用TLC检测仍有原料点。将反应液于70℃真空旋转蒸发后得到油状液体,后采取其他办法仍未能得到固体产物。可能是因为在酸性条件下,生成的产物与冰醋酸发生结合形成了阳离子,后处理过程较为复杂,难以分离。而在适量盐酸中反应数小时,也未见有产物生成。盐酸酸性过强,使反应无法进行;极大的可能是由于生成的单杂环二氮艹卓七元环环张力大,具有不稳定性,在强酸刺激下发生了开环,致使无法得到相应的产物。
在中性条件下,反应中有固体析出,TLC监测原料点逐渐变淡至消失,经抽滤、重结晶等简单后处理即可得到相应的目标产物。由此可见,在冰醋酸或盐酸等酸性条件下均不能高效地得到目标产物,而在中性条件却能够较好的得到目标产物。各反应物在中性反应体系下的实验结果见表1。

表1 中性条件下3a~3g的收率*Table 1 Yields of 3a~3g under neutral condition
2.2原料比r[r=n(2)∶n(1)]
1 30 mmol,其余反应条件同1.2,考察r对产物收率的影响,结果见图1。从图1可见,r对产物收率的影响呈规律性变化;随着r的增大,产物收率也明显增加;当r在1.2~1.4时,产物收率可达到各自相应的最高值。r继续增大,产物收率会随之下降。可能的原因是增加2的量后,2无法全部溶解于乙醇溶液中,并且2呈碱性,增加其用量使反应体系呈碱性,不利于反应进行,致使产物的收率下降。r在1.4时,2的投入量较1.2多,且2的沸点较高,若采用旋蒸法除尽2,温度须达180℃,而目标产物为不稳定的二氮七元杂环,在此高温下极可能开环。再者,2很容易与空气中的CO2反应生成不溶性盐混于目标产物中,2过量太多会增加后处理的难度,影响目标产物的收率。为此,最佳的r=1.2。
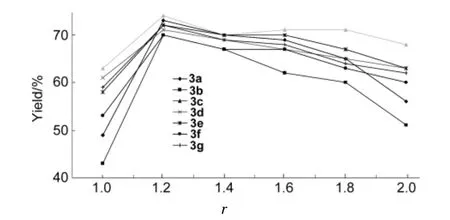
图1 r对产物收率的影响*Figure 1 Effect of r on yield of products*1 30 mmol,其余反应条件同1.2
2.3重结晶溶剂
以3a为例,对重结晶溶剂进行考察,结果见表2。由表2可以看出,3a在三氯甲烷、乙醇中加热均可溶解,分别进行重结晶后发现,三氯甲烷对3a的溶解度较大,不利于产物的析出,而在乙醇溶剂中加热有较好的溶解度,常温下大量析出,可得到较高纯度的3a。故本文选择乙醇为溶剂重结晶溶剂。
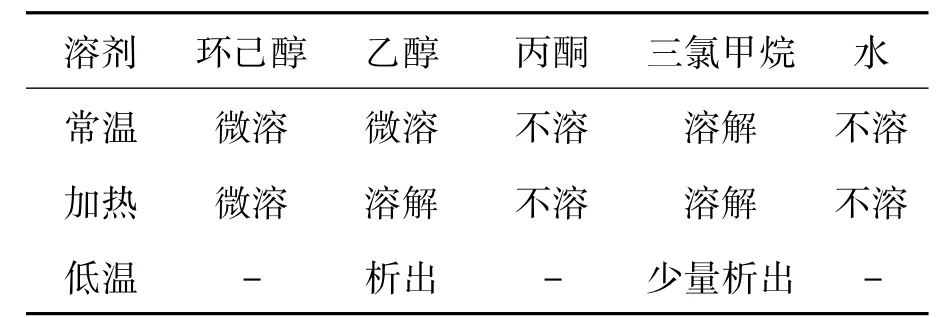
表2 3a的溶解性Table 2 Solubleness of 3a in different solvents
综上所述,合成3的最佳反应条件为:1 30 mmol,n(2)∶n(1)=1.2,回流反应8 h~10 h,无水乙醇为重结晶溶剂。
参考文献
[1]Wang X J,Tian Y L,Zhang Q Y,et al.An efficient synthesis of substituted l,4-diazepines by a Pd catalyzed a ination and sequential hydrogenation condensation[J].Chinese Chemical Letters,2013,4:743-746.
[2]苏杭,何西平,张丽,等.4-正丁基-3,4-二氢-1H-噻吩并[2,3-e][1,4]-二氮杂艹卓-2,5-二酮的合成[J].湖北大学学报(自然科学版),2011,33(4):501-504.
[3]靳颖华,史峻义.非苯二氮类镇静催眠药的研发进展与临床评价[J].中国医院用药评价与分析,2005,5(2):91-93.
[4]张蕾萍,舒翠霞,杜鸿雁.ASE和GPC方法检验非苯二氮艹卓类催眠药[J].刑事技术,2012,3:6-8.
[5]李斌,李剑峰,黄钢.咪唑并吡啶类外周苯二氮艹卓受体配体的合成新方法[J].上海第二医科大学学报,2005,25(12):1242-1245.
[6]王兰芝,花中霞,牛文刚.1,5-苯并二氮杂艹卓衍生物的合成及其生物活性的研究进展[J].有机化学,2010,30(11):1664-1671.
[7]Witt A,Gustavsson A,Bergman J,et al.Studies towards the synthesis of the benzdiazepin alkaloid auranthine:Synthesis of an actylated derivative[J].J Heterocyclic Chem,2003,40:29-35.
[8]王瑞霞,魏兆甫.失眠的用药选择[J].当代医学,2010,16(3):87-88.
[9]张石革.非苯二氮类催眠药作用靶位的特异性与评价[J].中国医院用药评价与分析,2006,6(2):72-74.
[10]Hassanien A Z,Ghozlan S A S,Elnagdi M H.Enaminones as building blocks in organic synthesis:A novel route to polyfunctionally substituted benzonitriles,pyridines,eneylbenzotriazoles and diazepines [J].J Heterocyclic Chem,2003,40:225-228.
[11]Fathi A A S,Sherif M S,Sayed A S M.Dimethylformamide dimethyl acetal as a building block in heterocyclic synthesis[J].J Heterocyclic Chem,2009,46:
通信联系人:刘飞,教授,Tel.0415-3789989,E-mail:berylliu8090@ sina.com
Synthesis of 5-Substituted-2,3-dihydro-1H-[1,4]diazepine Compounds
CHEN Qi-fan1,ZHANG Hui-dong1,WU Xiao-yan1,2,LIU Fei1
(1.School of Chemical Engineering,Eastern Liaoning University,Dandong 118003,China; 2.College of Chemistry,Northeast Normal University,Changchun 110004,China)
Abstract:Seven 5-substituted-2,3-dihydro-1H-[1,4]diazepine compounds(3a~3g,3a~3d were novel compounds)were synthesized by Michael nucleophilic addition reaction of enaminone(1)with ethylene diamine(2)in ethanol.The structures were characterized by1H NMR,IR and elemental analysis.The yields of 3a~3g were 70.0%~74.2% under the optimum reaction conditions[1 30 mmol,n(2)∶n(1)=1.2,reflux 8 h~10 h,absolute alcohol as recrystallization solvent].
Keywords:enaminone; Michael nucleophilic addition reaction;[1,4]diazepine; synthesis
作者简介:陈启凡(1964-),女,汉族,辽宁铁岭人,博士,主要从事有机合成的研究。Tel.0415-3789960,E-mail:qifan_c405@163.com
基金项目:辽宁省科技厅自然科学基金优秀人才培育项目(2014020172);辽宁省教育厅科学研究项目(L2013501)
收稿日期:2015-08-31
DOI:10.15952/j.cnki.cjsc.1005-1511.2015.12.1171 *
文献标识码:A
中图分类号:O626.5
