3-羟基-28-羧基熊果酸衍生物的合成及抗肿瘤活性研究
2016-01-12张良锋孟艳秋
张良锋, 孟艳秋
(沈阳化工大学 制药与生物工程学院, 辽宁 沈阳 110142)
熊果酸(ursolic acid,UA)又名乌索酸,是从枇杷叶、女贞叶和夏枯草等天然植物中分离得到的一种具有乌苏烷型骨架的五环三萜类天然产物[1-2].研究表明,熊果酸具有多种重要的生物活性,如:镇静、抗炎、抗菌、抗癌、抗溃疡、抗病毒、降低血糖、抗疟等作用[2-3].MA C M等[4]将熊果酸C-3位羟基烷酰化、C-11位氧化和C-28位羧基成酯,得到3-烷酰氧基-11-氧代-28-羧酸酯类熊果酸衍生物,具有很强的抗肿瘤活性.白育军等[5]将熊果酸C-3位氧化、C-28位羧基成酯得到熊果酸衍生物,具有很强的体外抗肿瘤活性.Gayathri C等[6]将熊果酸C-2位引入吸电子基团,如:I、CN和CF3,C-28位羧基成酰胺,得到的熊果酸衍生物具有很强的抗肿瘤作用.在五环三萜类化合物结构修饰的过程中,对羧基的修饰主要为羧基成酯和成酰胺的反应[7-10].本文以熊果酸(UA)为先导化合物,在本课题组前期对五环三萜类化合物结构改造以及构效关系研究的基础上[11-15],保留五环三萜骨架,对其C-3和C-28位进行结构修饰,合成了11个新的熊果酸衍生物,并对化合物进行了体外抗肿瘤活性的研究.
1 实验部分
1.1 仪器与试剂
Büchi B-540熔点测定仪;BrukerARX-500型核磁共振分析仪,CDCl3为溶剂,TMS为内标;热电-菲尼根LCQ型质谱仪;EA3000有机元素分析仪;Büchi R-200旋转蒸发仪;SHZ-2000型双配套循环水式多用真空泵;SANYO MC0175型CO2培养箱;SW-CJ-2FD型双人单面净化工作台;Olympus IX70倒置显微镜;BIO-RAD MODEL680型酶标仪;DL92125-125MM游标卡尺.
薄层色谱硅胶GF254,显色剂为体积分数为10 %的硫酸乙醇溶液;活性测试所用的培养基为RPMI-1640(含质量分数为10 %的胎牛血清,100 U·mL-1青霉素,质量浓度为100 mg·L-1的链霉素);溴化四氮唑盐(MTT)、蛋白酶K(Proteinase K)和小牛血清蛋白(BSA);HeLa细胞、HepG2细胞和BGC-823细胞由中国医学科学院协和医科大学药物研究所药理室提供.所有试剂均为分析纯或化学纯.
1.2 目标化合物的合成路线
目标化合物的合成路线如图1 所示.
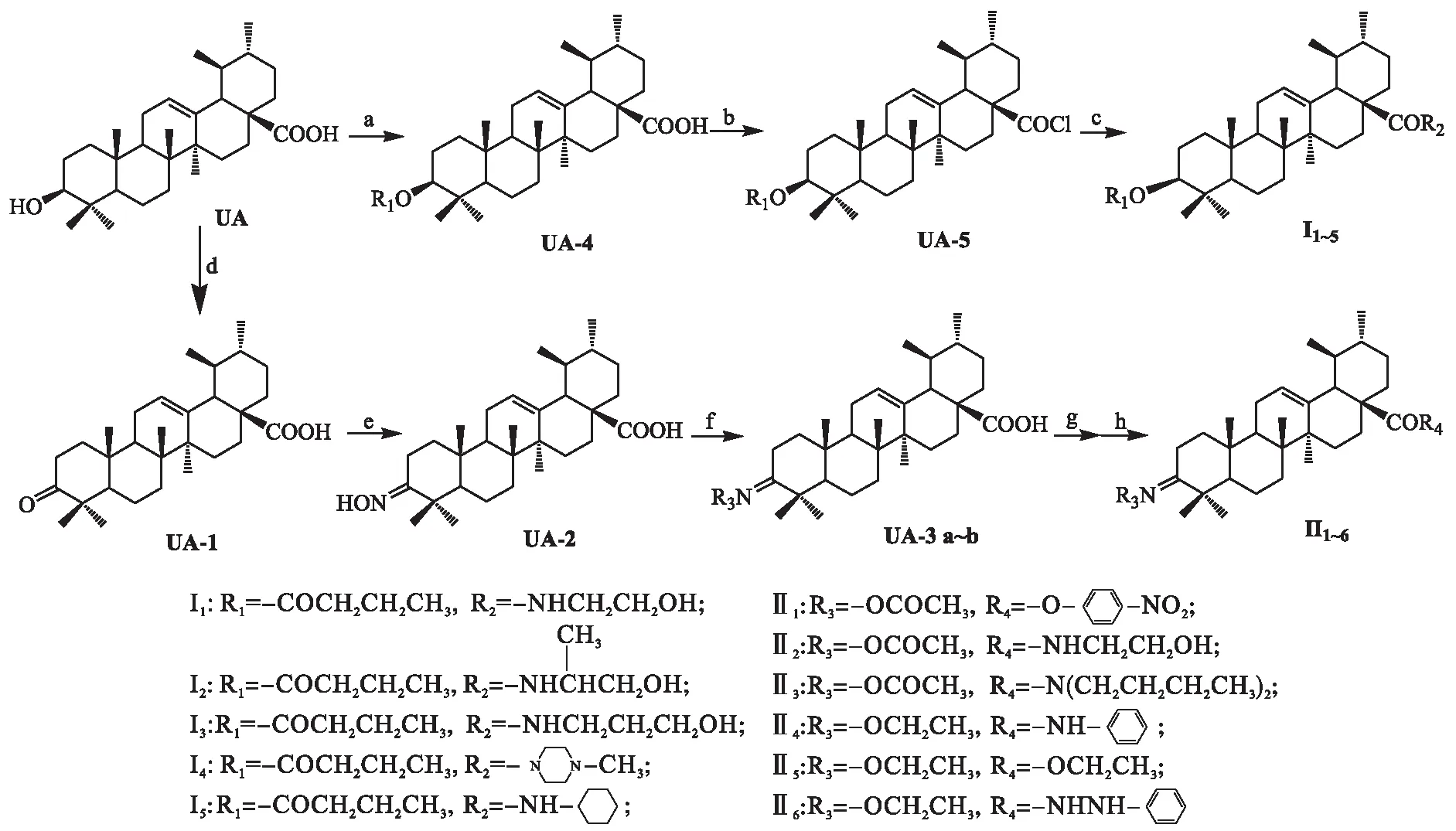
反应条件:a.DCC,DMAP,THF; b.(COCl)2,CH2Cl2; c.amines,CH2Cl2,rt; d.Jone′s reagent; e.NH2OH·HCl,Py;
f.Ac2O,Py,DMAP or EtBr,KOH,DMF; g.(COCl)2,CH2Cl2; h.amines or alcohol or phenol,CH2Cl2,rt
图1 目标化合物的合成路线
Fig.1 Synthetic routes of target compounds
1.3 中间体的合成
3-氧代熊果酸(UA-1):参照文献[1]的方法,制备化合物3-氧代熊果酸(UA-1),得白色固体35.4 mg,收率70.9 %.
3-肟基熊果酸(UA-2):将UA-1(50.0 mg,0.11 mmol)溶于适量吡啶中,加入盐酸羟胺(100 mg,1.44 mmol),115 ℃下回流1.5 h,反应完毕后倒入冰水中,产生大量白色沉淀,抽滤,水洗滤饼至中性,得白色固体42.8 mg,收率为83.0 %.
3-乙酰氧亚氨基-乌苏烷型-12-烯-28-酸(UA-3a):将UA-2(50.0 mg,0.11 mmol)溶于5 mL四氢呋喃溶液中,依次加入0.3 mL吡啶、乙酸酐(337 mg,3.30 mmol)和少量DMAP,室温下搅拌,TLC检测反应终点(展开剂为V(石油醚)/V(乙酸乙酯)=3/1,显色剂为体积分数10 % 的硫酸乙醇溶液),反应完毕,减压蒸除溶剂,加入3 mL水,用浓度为2 mol·L-1的盐酸调pH值至3~4,抽滤,滤饼水洗至中性,室温自然干燥,所得粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=10/1,得白色固体40.2 mg,产率为73.8 %,m.p. 87.5~91.4 ℃.
3-乙氧亚氨基-乌苏烷型-12-烯-28-酸(UA-3b):将UA-2(50.0 mg,0.11 mmol)溶于5 mL DMF中,依次加入1 mL溴乙烷和氢氧化钾(50.0 mg,0.89 mmol),70 ℃下搅拌1 h,TLC检测反应终点(展开剂:V(石油醚)/V(乙酸乙酯)=3/1,显色剂为体积分数10 %的硫酸乙醇溶液),加入适量二氯甲烷稀释,用浓度为2 mol·L-1的盐酸洗涤2次,饱和氯化钠水洗至中性,合并有机相,干燥,浓缩,所得粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=10/1,得白色固体36.2 mg,产率为68.4 %,m.p. 155.5~157.4 ℃.
3β-丁酰氧基-乌苏烷型-12-烯-28-羧酸(UA-4):参照文献[1]的方法,制备化合物3β-丁酰氧基-乌苏烷型-12-烯-28-羧酸(UA-4),得白色固体92.9 mg,产率80.2 %,m.p. 183.6~184.7 ℃.
1.4 目标化合物的合成
N-[3β-丁酰氧基-乌苏烷型-12-烯-28-酰]-2-氨基乙醇(Ⅰ1):将化合物UA-4(80.0 mg,0.15 mmol)溶于5 mL二氯甲烷中,缓慢滴加1 mL草酰氯,室温搅拌24 h,减压蒸除溶剂,加入3 mL环己烷,溶液呈浑浊状态,蒸干溶剂,重复该操作3次,得到化合物UA-5.将化合物UA-5溶于3 mL二氯甲烷中,加三乙胺调pH为9~10,搅拌5 min后加入2-氨基乙醇(37.1 mg,0.61 mmol),室温搅拌,TLC检测反应终点.反应完毕,向反应液中加入3 mL水,用浓度为2 mol·L-1的盐酸调pH至3,析出白色固体,抽滤,水洗滤饼至中性,室温干燥,粗品经硅胶柱色谱分离纯化,洗脱剂为V(石油醚)/V(丙酮)=5/1,得白色粉末状固体59.3 mg,产率70.3 %,m.p. 104.7~106.5 ℃.
N-[3β-丁酰氧基-乌苏烷型-12-烯-28-酰]-2-氨基-1-丙醇(Ⅰ2):按照化合物Ⅰ1的合成方法,由化合物UA-4(80.0 mg,0.15 mmol)与2-氨基-1-丙醇(45.6 mg,0.61 mmol)反应,所得粗品经硅胶柱色谱分离纯化,洗脱剂为V(石油醚)/V(丙酮)=3/1,得白色粉末状固体44.0 mg,产率50.8 %,m.p. 210.5~212.8 ℃.
N-[3β-丁酰氧基-乌苏烷型-12-烯-28-酰]-3-氨基丙醇(Ⅰ3):按照化合物Ⅰ1的合成方法,由化合物UA-4(80.0 mg,0.15 mmol)与3-氨基-1-丙醇(45.6 mg,0.61 mmol)反应,所得粗品经硅胶柱色谱分离纯化,洗脱剂为V(石油醚)/V(丙酮)=3/1,得白色粉末状固体36.3 mg,产率42.0 %,m.p. 210.7~212.3 ℃.
N-[3β-丁酰氧基-乌苏烷型-12-烯-28-酰]-4′-甲基哌嗪(Ⅰ4):按照化合物Ⅰ1的合成方法,由化合物UA-4(80.0 mg,0.15 mmol)与N-甲基哌嗪(60.9 mg,0.68 mmol)反应,所得粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(丙酮)=3/1,得白色粉末状固体55.7 mg,产率60.2 %,m.p. 82.6~84.3 ℃.
N-[3β-丁酰氧基-乌苏烷型-12-烯-28-酰]-环己胺(Ⅰ5):按照化合物Ⅰ1的合成方法,由化合物UA-4(80.0 mg,0.15 mmol)与环己胺(60.3 mg,0.61 mmol)反应,所得粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(丙酮)=10/1,得白色粉末状固体62.3 mg,产率67.5 %,m.p. 115.3~117.5 ℃.
3-乙酰氧亚氨基-乌苏烷型-12-烯-28-酰-4′-硝基苯酚酯(Ⅱ1):将化合物UA-3a(50.0 mg,0.10 mmol)溶于5 mL二氯甲烷中,缓慢滴加2 mL草酰氯,室温搅拌20 h,减压蒸除溶剂,加入3 mL环己烷,蒸干溶剂,重复该操作3次,得到3-乙酰氧亚氨基-乌苏烷型-12-烯-28-酰氯.将得到的酰氯溶于3 mL二氯甲烷中,加三乙胺调pH至 9 ~10,搅拌5 min后,加入对硝基苯酚(20.9 mg,0.15 mmol),室温搅拌,TLC检测反应终点.减压蒸除溶剂,加入2 mL水,用浓度为2 mol·L-1的盐酸调pH至 3~4,析出白色固体,抽滤,水洗滤饼至中性.所得粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=15/1,得白色粉末状固体24.5 mg,产率39.1 %,m.p. 104.2~106.1 ℃.
N-[3-乙酰氧亚氨基-乌苏烷型-12-烯-28-酰]-2-氨基乙醇(Ⅱ2):按照化合物Ⅱ1的合成方法,由化合物UA-3a(50.0 mg,0.10 mmol)经酰氯与2-氨基乙醇(35.0 mg,0.57 mmol)反应,所得粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=8/1,得白色粉末状固体18.7 mg,产率34.1 %,m.p. 90.7~91.3 ℃.
N-[3-乙酰氧亚氨基-乌苏烷型-12-烯-28-酰]-二丁胺(Ⅱ3):按照化合物Ⅱ1的合成方法,由化合物UA-3a(50.0 mg,0.10 mmol)经酰氯与3 mL二丁胺反应,所得粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=10/1,得白色粉末状固体19.0 mg,产率31.8 %,m.p. 106.3~107.5 ℃.
N-[3-乙氧亚氨基-乌苏烷型-12-烯-28-酰]-苯胺(Ⅱ4):按照化合物Ⅱ1的合成方法,由化合物UA-3b(50.0 mg,0.10 mmol)经酰氯与2 mL苯胺反应,所得粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=10/1,得白色固体34.5 mg,产率为59.95 %,m.p. 142.5 ~144.3 ℃.
3-乙氧亚氨基-乌苏烷型-12-烯-28-羧酸乙酯(Ⅱ5):将化合物UA-3b(50.0 mg,0.10 mmol)和无水碳酸钾(30 mg,0.22 mmol)加到5 mL DMF中,然后加入2滴溴乙烷,室温搅拌,TLC检测反应终点.加入2 mL饱和食盐水,用乙酸乙酯萃取3次,合并有机相,水洗至中性,浓缩,干燥,所得粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=9/1,得白色粉末状固体18.5 mg,产率35.2 %,m.p. 169.5~170.4 ℃.
N-[3-乙氧亚氨基-乌苏烷型-12-烯-28-酰]-苯肼(Ⅱ6):按照化合物Ⅱ1的合成方法,由化合物UA-3b(50.0 mg,0.10 mmol)经酰氯与苯肼(16.2 mg,0.15 mmol)在室温条件下反应,所得粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=10/1,得白色粉末状固体22.4 mg,产率38.1 %.m.p. 178.4~180.6 ℃.
1.5 初步体外细胞毒活性测试
以吉非替尼为阳性对照药,采用MTT法对合成的目标化合物进行体外抗肿瘤活性测试.所选用的肿瘤细胞为人宫颈癌细胞(HeLa)、人肝癌细胞(HepG2)和人胃癌细胞(BGC-823).对数生长期肿瘤细胞培养于96孔培养板内,每孔100 μL(大约含4000个细胞),置37 ℃、摩尔分数为5 %的CO2温箱中培养.培养24 h后,给药组加入浓度为1×10-5mol·L-1的测试物,至少设3个平行孔,阴性对照组加入与给药组等体积的溶剂,并设只加培养基的空白对照组,置37 ℃、摩尔分数为5 %的CO2温箱中培养.48 h后弃培养液,每孔加入MTT溶液(质量浓度为1 g·L-1)50 μL,在37 ℃下,孵育4 h,弃上清液,每孔加入150 μL DMSO溶解甲臜颗粒,轻度振荡溶解.在酶标仪波长490 nm条件下测定各孔光密度值(OD),计算所测化合物对细胞的抑制率和IC50值.试验重复3次,结果取平均值.
抑制率=

2 结果与讨论
2.1 化学合成
以熊果酸(UA)为起始原料,经过酰化、酯化或酰胺化等反应得到11个目标化合物,目标化合物的结构经1H-NMR、元素分析和MS确定,均为新化合物,数据见表1和表2.

表1 目标化合物的质谱和元素分析数据
注:括号内为计算值

表2 目标化合物的氢谱数据
2.2 抗肿瘤活性测试
以吉非替尼为阳性对照药,采用MTT法测试目标化合物体外抗肿瘤活性,活性数据见表3.结果表明:所测试的目标化合物对HeLa、HepG2和BGC-823三种细胞生长均具有一定的抑制作用,其中化合物Ⅱ4和Ⅱ6对三种肿瘤细胞均表现出很强的抑制活性.化合物Ⅱ4对HeLa、HepG2和BGC-823细胞的抑制率分别为50.3 %、59.8 %和62.1 %,明显高于临床常用药物吉非替尼.化合物Ⅱ6对HeLa、HepG2和BGC-823细胞的抑制率分别为44.7 %、45.0 %和50.1 %,明显高于母体化合物UA,与吉非替尼相当.可以看出:C-3位羟基氧化后成肟,引入烷氧亚氨基,同时在C-28位引入酰胺基团可以提高其抗肿瘤活性.化合物Ⅱ4和Ⅱ6对三种肿瘤细胞的抑制活性明显高于Ⅰ1和Ⅱ3等化合物.所以,C-28位羧基所连接胺的结构对抑制活性可能有较大的影响.

表3 目标化合物对HeLa、HepG2和BGC-823肿瘤细胞增殖的抑制活性
注:化合物浓度为1×10-5mol·L-1,经过72 h处理后的细胞抑制率; nt:not tested.
3 结束语
初步的药理实验结果表明:C-3位引入烷氧亚氨基(或羟基成酯),C-28位羧基成酯或酰胺,对其体外抗肿瘤活性具有提高作用.可见,合理的修饰熊果酸C-3位羟基、C-28位羧基,对熊果酸抗肿瘤活性有较大影响.这对下一步的结构设计和优化具有一定的指导意义,为今后研究和开发高效低毒的抗肿瘤药物打下良好的基础.
[1] MENG Y Q,LIU D,BAI Z W,et al.Synthesis and Anti-tumor Activity of Ursolic Acid Derivatives[J].Acta Pharmaceutica Sinica,2011,46(5):556-560.
[2] 夏燕,孟艳秋,仇兴华,等.熊果酸A环开环结构修饰工艺路线的设计[J].中草药,2011,42(1):34-37.
[3] 赵龙铉,杨君微,郑昌吉,等.熊果酸衍生物的合成与表征及其抑菌活性研究[J].辽宁师范大学学报(自然科学版),2012,35(3):358-363.
[4] MA C M,CAI S Q,CUI J R,et al.The Cytotoxic Activity of Ursolic Acid Derivatives[J].European Journal of Medicinal Chemistry,2005,40(6):582-589.
[5] 白育军,杨小生,康文艺,等.熊果酸的结构修饰物及其抗肿瘤活性[J].华西药学杂志,2003,18(2):87-90.
[6] CHADALAPAKA G,JUTOORU I,McALEES A,et al.Structure-dependent Inhibition of Bladder and Pancreatic Cancer Cell Growth by 2-substituted Glycyrrhetinic and Ursolic Acid Derivatives[J].Bioorganic & Med.Chem.Lett.,2008,18(8):2633-2639.
[7] 申利红,赖宜生,张奕华.硝酸酯类甘草次酸衍生物的合成及抗肿瘤活性[J].中国药科大学学报,2008,39(2):103-107.
[8] 孟艳秋.乳香酸类化合物的结构改造及其抗癌活性的研究[D].沈阳:沈阳药科大学,2004:52-67.
[9] 孟艳秋,刘丹,李丹丹,等.一种具有抗肿瘤活性的熊果酸化学修饰物胺:CN101161670B[P].2008-04-16.
[10] ZHAO L X,PARK H G,JEW S S,et al.Modification of C11,C28,C2,3,23 or C2,23,28 Functional Groups on Asiatic Acid and Evaluation of Hepatoprotective Effects[J].Cheminform,2007,38(45):970-976.
[11] MENG Y Q,NIE H H,WANG X C,et al.Synthesis and Anti-tumor Activity of Oleanolic Acid Derivatives[J].Acta Pharma Sinica,2011,46(10):1215-1220.
[12] MENG Y Q,LIU D,CAI L L,et al.The Synthesis of Ursolic Acid Derivatives with Cytotoxic Activity and the Investigation of Their Preliminary Mechanism of Action[J].Bioorg Med Chem,2009,17(2):848-854.
[13] LIU D,MENG Y Q,ZHAO J,et al.Synthesis and Anti-tumor Activity of Novel Amide Derivatives of Ursolic Acid[J].Chem Res Chin Univ,2008,24(1):42-46.
[14] MENG Y Q,SONG Y L,YAN Z K,et al.Synthesis and in Vitro Cytotoxicity of Novel Ursolic Acid Derivatives[J].Molecules,2010,15(6):4033-4040.
[15] 孟艳秋,薛菁,刘凤鑫,等.齐墩果酸衍生物的合成及抗肿瘤活性研究[J].化学通报,2013,76(3):265-269.
