药用中间体1-甲基-5-巯基-1H-四氮唑相转移合成工艺研究
2016-01-11常相娜张健宇宁肖肖
常相娜, 张健宇, 宁肖肖, 沈 文
(1.陕西科技大学 生命科学与工程学院, 陕西 西安 710021; 2.杨凌示范区 食品药品检验所, 陕西 杨凌 712100)
药用中间体1-甲基-5-巯基-1H-四氮唑相转移合成工艺研究
常相娜1, 张健宇2, 宁肖肖1, 沈文1
(1.陕西科技大学 生命科学与工程学院, 陕西 西安710021; 2.杨凌示范区 食品药品检验所, 陕西 杨凌712100)
摘要:在水相中,相转移催化剂催化条件下合成1-甲基-5-巯基-1H-四氮唑,并对其工艺条件进行研究.采用IR与1HNMR对产物结构进行了表征.结果表明催化剂的最优制备条件为:催化剂为环糊精、蒸馏水加入量为30 mL、催化剂加入量为1%(占叠氮钠的摩尔百分数).在此催化条件下优化工艺条件为:异硫氰酸甲酯与叠氮钠的摩尔比为1.4∶1.0;滴加完异硫氰酸甲酯后的反应时间为2 h;反应温度为75 ℃.产物收率为76.93%.
关键词:1-甲基-5-巯基-1H-四氮唑; 相转移催化; 工艺
0引言
四唑类结构作为有机合成中间体在医药、含能材料、农业、配位聚合物制备及固体催化剂等领域有着广泛的应用[1-5].四唑结构的主要合成方法是叠氮酸、叠氮离子及叠氮有机物对碳氮多重键的加成反应.传统合成工艺多数使用有机溶剂系统,并采用金属化合物进行催化,存在着环境友好性较差、合成成本高、不利于产业化的不足.如:合成 1,5-二取代四唑的文献报道大多是利用亚酰胺或硫代酰胺与 PCl5/HN3,TMSN3/Ph3P/DEAD(Diethyl azodicarboxylate),TMSN3/Et3N/Hg(II) (合成路线如图1所示)等反应合成1,5-二取代四唑.这些反应条件比较苛刻,收率低,后处理繁琐,催化剂有毒性,因此限制了它们在工业生产等方面的应用[6].因此发展清洁、经济和环境较友好条件下的四唑类有机合成方法引起有机合成工作者的关注,其中,相转移催化反应、无溶剂反应、微波反应非常具有发展潜力[7-12].本文在已有的研究基础上[13-17]深入研究相转移催化合成1-甲基-5-巯基-1H-四氮唑(MMT)的工艺,为相转移催化合成四唑类结构研究提供参考.

图1 合成路线
1实验部分
1.1仪器材料
异硫氰酸甲酯(沃凯,≥98%),叠氮钠(天津市天力化学试剂有限公司,化学纯),MMT对照品(sigma,98%),其余试剂均为分析纯,水为去离子水.
主要仪器:WRS-2A微机熔点仪(上海精密仪器有限公司);VECTOR-22傅立叶红外光谱仪(德国BRUKER公司);400 MHz核磁共振波谱仪(德国BRUKER公司);1 200LC高效液相色谱仪(美国Agilent公司).
1.2合成与表征
1-甲基-5-巯基-1H-四氮唑合成路线如图2所示.

图2 1-甲基-5-巯基-1H-四氮唑合成路线
将叠氮钠2.96 g,去离子水30 mL加入到连有搅拌器和回流冷凝管的三口烧瓶中,再加入相转移催化剂并通过恒温水浴锅保持体系在75 ℃.将4.0 g异硫氰酸甲酯溶解在10 mL无水乙醇中,通过恒压滴液漏斗滴加进入反应体系,1 h加完.在此温度下搅拌反应2 h,然后将物料浓缩,降温至10 ℃下,用浓盐酸酸化至pH=2,然后降温结晶,过滤得甲基巯基四氮唑粗品.将得到的甲基巯基四氮唑粗品加入少量去离子水溶解,再用质量分数为25%~28%的氨水溶液调节pH至8~9,过滤,加入盐酸溶液调节pH约为2,结晶、过滤.氯仿重结晶得甲基巯基四氮唑精品.所得产物为白色结晶,有刺激气味.收率76.93%. m.p.125.1 ℃~126.2 ℃(文献值:125 ℃~128 ℃).纯度≥95.0%.1H NMR (400 MHz, D2O)δ:4.79(s,1H,HS),3.76(s,3H,CH3).IR( KBr压片):四唑环内的C-N、N-N、C=N、N=N的伸缩振动 (1 195~1 600 cm-1)[17],其中C=N伸缩振动(1 510 cm-1),C-N伸缩振动(1 213 cm-1),N-N的吸收峰(1 043 cm-1);C-H的伸缩振动(2 919 cm-1、2 858 cm-1);C-H的弯曲振动(1 432 cm-1、1 380 cm-1);S-H的伸缩振动(2 068 cm-1);C-S的吸收峰(669 cm-1).
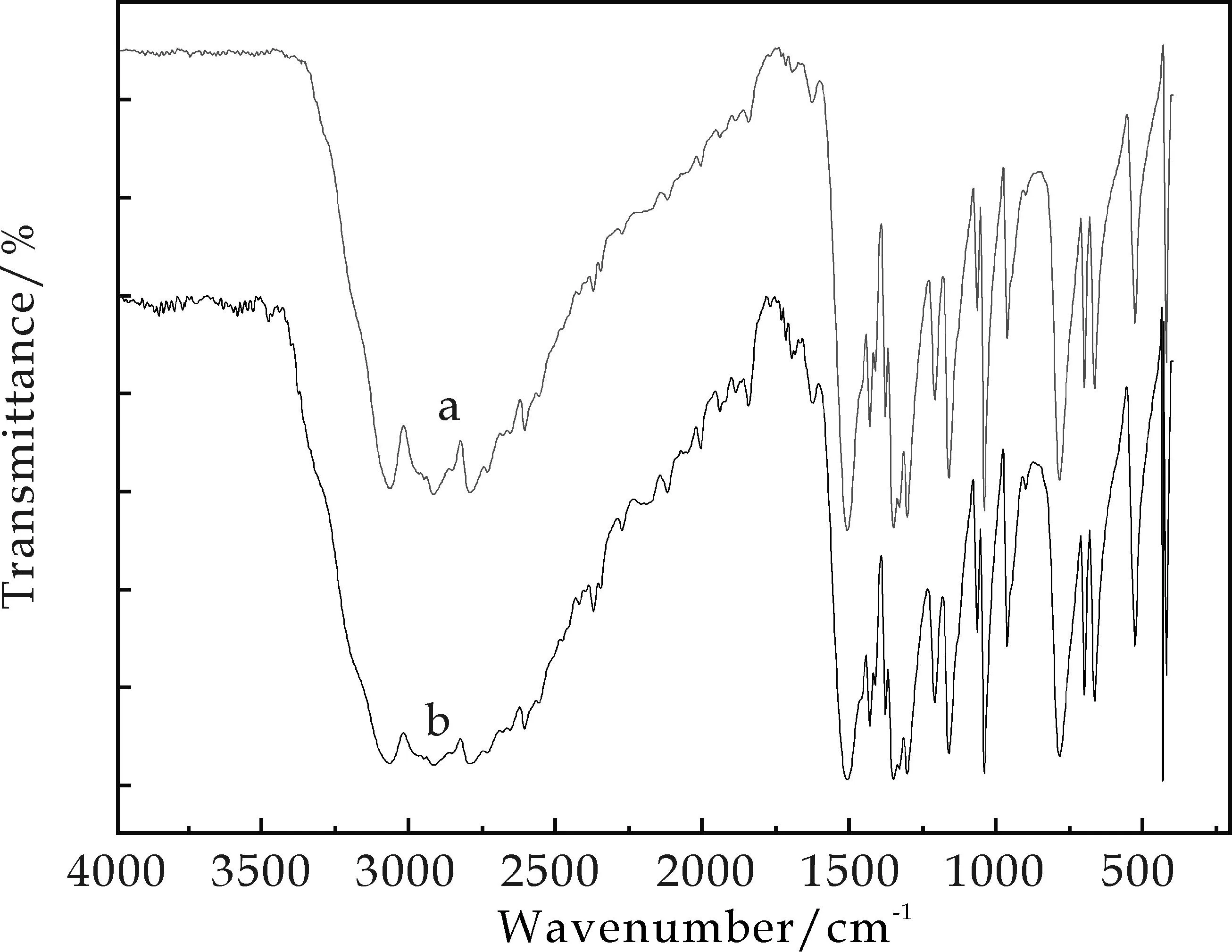
a:对照品;b:样品图3 MMT对照品a与样品b的红外图谱
1.3分析方法
1.3.1分析条件
色谱柱:Diamonsil C18,150 mm×4.6 mm×5μm;检测器:UV254nm;柱温:室温;流动相:VA∶VB∶V缓冲溶液1∶V缓冲溶液2=550∶440∶55∶5;A:乙腈;B:水;缓冲溶液l:称取磷酸氢二钾13.6 g和磷酸二氢钾4.0 g,溶于1 000 mL水中,调节pH到7.0.缓冲溶液2:称取柠檬酸钠25.8克,置1 000 mL容量瓶中,加水500 mL使溶解,用20%柠檬酸溶液调节pH值5.0,加水至刻度;流速:0.8 mL/min,进样量:5μL;外标法定量.
1.3.2标准曲线
用精密称取MMT对照品100 mg,用流动相溶解并定容于100 mL容量瓶中,得到质量浓度为1 mg/mL的母液.分别从母液中移取1、2、3、4、5、6、7、8、9 mL置于10 mL量瓶中,流动相定容至刻度,得到质量浓度分别为0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9 mg/mL系列对照品溶液.精密吸取5μL上述对照品溶液,进行测定.由MMT峰面积(Y)对质量浓度(X)进行线性回归,得回归方程Y=1 668.4X+262.14,R=0.999 05,表明MMT在0.1~1 mg/mL浓度范围内线性关系良好.
1.3.3含量测定
精密称取样品约4 mg,置于10 mL容量瓶中,用流动相溶解并定容.摇匀,精密量取5μL进行测定,样品保留时间为3.9 mim左右,样品运行时间应为主峰保留时间的2倍以上,由测定得到的峰面积按标准曲线法计算纯度.产物的纯度≥95.0%.
2工艺优化结果与讨论
2.1相转移催化剂催化性能研究结果与讨论
通过对文献和相转移催化的研究,确定相转移催化影响的主要因素,A为相转移催化剂种类、B蒸馏水加入量(mL)、C催化剂加入量(占叠氮钠的摩尔百分数),这其中选取了PEG2000(链状聚乙二醇类)、环糊精(环状冠醚类)与四丁基溴化铵(季铵盐类)作为反应相转移催化剂.按L9(34)正交试验表设计实验方案,以收率为考察指标,从而对不同类型相转移催化剂性能进行比较.正交试验设计及结果如表1所示,方差分析如表2所示.

表1 L9(34)正交试验方案与结果
通过直观分析和极差R值的比较,各因素对实验结果影响的大小顺序为C>A>B.方差分析结果表明,因素C对收率有极显著的影响(P<0.01),而A、B因素的影响较小.综合上述分析得到最佳相转移催化工艺条件为A2B2C2:即相转移催化剂为环糊精,蒸馏水加入量为30 mL,催化剂加入量为1.0(占叠氮钠的摩尔分数).最佳工艺验证后其产品收率为75.69%,产品的纯度为98.07%.

表2 正交试验方差分析表
F0.05(2,2)=19.00;F0.01(2,2)=99.00
2.2操作条件优化结果与讨论
2.2.1单因素优化水平的确定
在确定的相转移催化剂为环糊精、蒸馏水加入量为30 mL、催化剂加入量为1.0(占叠氮钠的摩尔分数)的条件下研究单因素变化趋势与水平.
(1)异硫氰酸甲酯与叠氮钠的摩尔比的最优水平
由图4可以看出,当异硫氰酸甲酯与叠氮化钠的摩尔比从1.1∶1.0增加1.3∶1.0时,收率迅速增加,但是当异硫氰酸甲酯与叠氮化钠的摩尔比继续增大时,收率增加的很慢.综合考虑对反应的影响程度和路线的经济性,选取投料比的因素水平为:异硫氰酸甲酯与叠氮化钠的摩尔比=1.2∶1.0和1.4∶1.0.

图4 不同的投料比对MMT收率的影响
(2)反应时间的最优水平的确定
从图5可以看出滴加完异硫氰酸甲酯后,反应时间从0.5 h增加到3 h,反应进行的收率也增加;当反应时间超过4 h后,收率下降.2 h和3 h的收率最高.本研究选取的反应时间水平为2 h和3 h.

图5 反应时间对MMT收率的影响
(3)反应温度的最优水平的确定
从图6可以看出,当反应温度从65 ℃增加到75 ℃时,收率也呈增加趋势:当温度从75 ℃增加到80 ℃时,收率呈下降趋势.温度为70 ℃和75 ℃时,收率最高.本研究选取的反应温度水平为70 ℃和75 ℃.

图6 反应温度对MMT收率的影响
2.2.2MMT工艺优化结果与讨论
依照单因素水平研究确定的因素水平,按L8(27)正交试验表设计实验方案,以收率为考察指标,从而对相转移催化MMT合成工艺条件进行研究.其中A因素为投料比(n异硫氰酸甲酯:n叠氮钠),B因素为反应时间(h),C因素为反应温度(℃),并同时研究了各因素之间的交互作用.相转移催化剂采用环糊精,蒸馏水加入量为30 mL,催化剂加入量为1.0(占叠氮钠的摩尔分数).正交试验设计及结果如表3所示,方差分析如表4所示.

表3 L8(27)正交试验方案与结果

表4 方差分析表

F0.05(1,2)=18.51;
综合极差分析与方差分析可知各因素实验结果影响大小顺序为:B×C>B>A>A×B>C>A×C,其中因素B、C因素的交互作用与因素B影响显著.因B、C因素的交互作用对实验结果的影响最显著,故结合考虑B、C因素的交互作用(如表5所示)确定最佳工艺条件为A2B1C2.最佳工艺验证后收率为76.93%,产品的纯度为99.52%.
反应过程中,在一定范围内异硫氰酸甲酯分批加料时间延长会提高MMT收率,可以使反应更充分的进行,得到很好的收率.这是因为分批加入减少了短时间内反应物异硫氰酸甲酯的过量,从而减少了副反应的反应,而如果超出范围继续延长时间,反应收率提高的较小.而为了安全起见,保证不会产生有剧毒的叠氮酸一定要让异硫氰酸甲酯部分过量.此外,温度在反应过程很重要,太低则达不到活化能,反应无法进行,只有达到一定温度才可以反应;温度高于80 ℃不仅会发生副反应,而且液体反应颜色极易变黄,且颜色不断加深,影响产物的外观和收率.
3结论
在相转移催化合成MMT反应中,催化剂的最优工艺条件为:催化剂为环糊精、蒸馏水加入量为30 mL、催化剂加入量为1%(占叠氮钠的摩尔百分数).在此催化条件下优化制备条件为:叠氮钠与异硫氰酸甲酯的摩尔比=1∶1.4;滴加完异硫氰酸甲酯后的反应时间为2 h;反应温度为75 ℃.实验所得产品,经过熔点测定、红外光谱、核磁质谱确证了结构.运用高效液相色谱法对含量进行了测定,产物的纯度≥95.0%,最高可达99.52%.
本合成工艺操作简单,产品纯度较高,且反应过程在水溶剂环境中进行,有利于降低生产成本,并避免了环境污染,因此,具有一定的经济效益和社会效益.
参考文献
[1] Taterao M.Potewar,Shafi A.Siddiqui,Rajgopal J.Lahoti,et al.Effcient and rapid synthesis of 1-substituted-1H-1,2,3,4-tetrazoles in the acidic ionic liquid 1-n-butylimidazolium tetrafiuoroborate[J].Tetrahedron Letters,2007,48(10):1 721-1 724.
[2] 张光全.绿色四唑类起爆药研究的最新进展[J].含能材料,2011,19(4):473-478.
[3] 魏太保,李伟,徐蓉,等.2-(1-苯基-1H-四唑-5-硫基)-N-芳基乙酰胺的合成及其生物活性研究[J].有机化学,2008,28(1):99-103.
[4] 王宝雷,李正名,李永红,等.4-烷氧/苄氧基基苯基四唑和1,3,4-噁二唑类化合物的合成及除草活性[J].高等学校化学学报,2008,29(1):93-94.
[5] 张苏杭,张玉清,王新德.四唑化合物的合成及其在农业上的应用[J].洛阳工业高等专科学校学报,2006,16(2):16-18.
[6] 李冠琼,李玉川,马巧丽,等.富氮唑环类化合物的环加成合成研究进展[J].有机化学,2010,30(10):1 431-1 440.
[7] 李伟,张有明,魏太保,等.微波作用下1-苯基-5-[5-(芳胺羰基甲硫基)-4-苯基-1,2,4-三唑-3-甲硫基]四唑的合成及生物活性[J].有机化学,2008,28(3):454-458.
[8] Mara Rubia Couri, Inacio Luduvico, Leandro Santos,et al.Microwave-assisted efficient preparation of novel carbohydrate tetrazole derivatives[J].Carbohydrate Research,2007,342(8):1 096.
[9] 王立中,张文华.水热法合成3-吡啶四唑的研究[J].山西化工,2004,24(3):22-23.
[10] 徐文龙,陈敏东,姜玲,等.利用叠氮化合物固相方法合成含氮杂环化合物的研究进展[J].化学试剂,2010,32(4):317-321.
[11] 黄家吉,贺晓鹏,董菁,等.利用分子内Click反应合成哌嗪并三唑和哌嗪并四唑化合物[J].合成化学,2010,18(1):64-66.
[12] 刘学东,柴生勇,鲁鸣久.1,5-二甲基四唑的合成研究[J].应用化工,2000,29(3):27-29.
[13] 梁晓琴.四唑衍生物结构及性质的理论研究[J].四川师范大学学报(自然科学版),2008,31(2):219-223.
[14] 李永伟,聂新永,田金如,等.绿色环保溶剂在 5-乙硫基-1H-四氮唑制备上的应用[J].化学世界,2007(8):478-479.
[15] 刘丽秀,鲁玲玲,王灏,等.甲基巯基四唑合成工艺研究[J].四川化工,2004,7(3):10-12.
[16] 冯维春,孟宪兴,王灏,等.1-甲基-5-巯基-1,2,3,4-四氮唑的生产方法[P].中国专利:201010115803.3,2010-07-21.
[17] 刘庆,甘孝贤,葛忠学,等.2-甲基-5-乙烯基四唑的合成与表征[J].火炸药学报,2010,33(5):30-32.

Synthesis of 5-mercapto-1-methyltetrazole
by phase transfer catalysis
CHANG Xiang-na1, ZHANG Jian-yu2, NING Xiao-xiao1, SHEN Wen1
(1.College of Life Science and Engineering, Shaanxi University of Science & Technology, Xi′an 710021, China; 2.Yangling Institute for Food and Drug Control, Yangling 712100, China)
Abstract:In the presence of phase-transfer catalyst,5-mercapto-1-methyltetrazole was synthesized in aqueous phase.The structure was characterized by means of IR and1H NMR.The optimum condition of phase transfer catalyst were ascertained as follows:the phase transfer catalyst was cyclodextrin, the quality of deionized water was 30 mL,and the quality ratio of phase transfer catalyst and sodium azide was 1∶100(mol/mol).Using cyclodextrin as phase transfer catalyst,the optimum preparation procedure was:reaction temperature was 70 ℃,the quality ratio of methyl isothiocyanate and sodium azide was 1.4∶1.0 (mol/mol), reaction time was 2 h, and the yield of 5-mercapto-1-methyltetrazole was 79.63%.
Key words:5-mercapto-1-methyltetrazole; phase transfer catalysis; technology
中图分类号:R97
文献标志码:A
文章编号:1000-5811(2015)01-0131-05
作者简介:常相娜(1977-),女,黑龙江伊春人,讲师,研究方向:药物合成及药用新材料
基金项目:陕西省科技厅自然科学基础研究计划项目(2011JM2005); 陕西省教育厅专项科研计划项目(2013JK0767)
收稿日期:*2014-11-24
