稀土掺杂对氯化氢氧化制氯气CuO-CeO2-SiO2催化剂结构和性能的影响
2015-12-29谢兴星费兆阳李郑州献汤吉海崔咪芬旭1
谢兴星 费兆阳 邹 冲 李郑州 陈 献汤吉海 崔咪芬 乔 旭1,,*
(1南京工业大学材料化学工程国家重点实验室,南京210009;2南京工业大学化学化工学院,南京210009)
稀土掺杂对氯化氢氧化制氯气CuO-CeO2-SiO2催化剂结构和性能的影响
谢兴星1,2费兆阳1,2,*邹 冲1,2李郑州1,2陈 献2汤吉海2崔咪芬2乔 旭1,2,*
(1南京工业大学材料化学工程国家重点实验室,南京210009;2南京工业大学化学化工学院,南京210009)
采用模板法制备了CuO-CeO2-SiO2和稀土掺杂的CuO-Ce0.9M0.1O2-SiO2(M=La,Pr,Nd)催化剂.运用X射线衍射(XRD),N2吸附-脱附,透射电镜(TEM),拉曼(Raman)光谱,X射线光电子能谱(XPS)和氢气-程序升温还原(H2-TPR)等手段对催化剂的结构进行表征,并考察稀土掺杂对氯化氢催化氧化制氯气性能的影响.结果表明,稀土掺杂进入CeO2晶格中形成良好的固溶体结构,获得更小的晶粒尺寸和更高的比表面积,并且显著提高了固溶体的表面氧空位浓度.稀土掺杂显著影响了催化剂的氯化氢催化氧化活性,活性顺序为:CuOCe0.9La0.1O2-SiO2>CuO-Ce0.9Nd0.1O2-SiO2>CuO-Ce0.9Pr0.1O2-SiO2>CuO-CeO2-SiO2,固溶体氧空位浓度的高低与氯化氢氧化活性直接相关.通过与Ce0.9M0.1O2-SiO2催化剂的结构和性能的对比,发现氧空位浓度的提高并不能增强在固溶体表面发生的氯化氢氧化反应.动力学测试显示,稀土掺杂后,氧分子的吸附成为反应过程的决速步骤.但在V(O2):V(HCl)=1条件下,更高的氧空位浓度导致了固溶体更低的氯化氢氧化反应速率.结合机理分析认为,CuO-Ce0.9M0.1O2-SiO2催化剂更高的氧空位浓度增强了固溶体表面的“氧溢流”,加快了氯化氢氧化的整体反应速率,这是CuO-Ce0.9M0.1O2-SiO2具备高活性的关键.
稀土;铈铜复合氧化物;氯化氢;催化氧化;氯气;氧空位
1 引言
氯气作为一种重要的基础化工原料,广泛应用于造纸、医药、纺织、石油化工、环保等行业.但是,以氯气为原料的许多生产过程会副产大量的氯化氢,以快速发展的聚氨酯和聚碳酸酯行业为例,全部氯元素都是以副产氯化氢的形式排出.1副产氯化氢如果不能被合理的利用,不仅会造成氯资源的极大浪费,同时还会对环境造成严重的污染.氯化氢通过催化氧化法制氯气,能够实现氯资源的循环利用,是具有广阔应用前景的绿色环保工艺.2-4
目前,住友和拜耳公司分别开发了RuO2催化剂,其具备较好的低温活性和催化稳定性,成功实现了催化氯化氢氧化制氯气的工业应用.5,6但是, RuO2催化剂存在成本昂贵,且活性温区窄,高温易流失等问题,限制了其进一步大规模使用.7因此,成本较低且具备高活性和稳定性的催化剂的开发已成为氯化氢氧化研究的热点.CeO2及铈基复合氧化物因其优异的储释氧性能而被广泛应用.8-10最近研究显示CeO2在较宽温区范围内具有较好的氯化氢氧化催化性能,被认为是可替代RuO2的新型氯化氢氧化催化剂.11以ZrO2为载体采用浸渍法制备了负载型CeO2/ZrO2催化剂,CeO2高度分散在ZrO2的表面形成了纳米薄层,显示出良好的氧化还原能力和抗氯化性能.12铜铁矿结构的CuCrO2与CeO2复合后氯化氢氧化催化活性较单组分提高四倍,而CeO2的氧空位的存在显著增强了其催化性能.13在前期的工作中,14我们采用Y分子筛为载体,制备出的CuOCeO2/Y催化剂在氯化氢氧化反应中表现出极高的活性和稳定性,具备良好的工业应用前景.详细的研究表明,15高分散的CuO为最主要的活性组分,CuO和CeO2之间存在着协同作用,铈铜固溶体的表面氧空位能够促进氧分子在催化剂表面的吸附和活化,这是催化剂具备高活性的关键.
稀土的掺杂可以有效改变CeO2的结构和物理化学性质,尤其是表面氧空位浓度和氧化还原性能.16,17然而,对于稀土掺杂对氯化氢氧化CuO-CeO2催化剂结构和性能影响的研究甚少.本文采用模板法制备了CuO-CeO2-SiO2和CuO-Ce0.9M0.1O2-SiO2(M= La,Pr,Nd)催化剂,通过多种表征手段考察稀土掺杂对催化剂的结构以及氯化氢氧化性能影响.同时还制备了Ce0.9M0.1O2-SiO2(M=La,Pr,Nd)催化剂,通过与其结构和性能的对比,并结合动力学手段,尝试阐述氯化氢氧化在CuO-Ce0.9M0.1O2-SiO2表面反应过程.
2 实验部分
2.1 催化剂制备
CuO-CeO2-SiO2催化剂采用模板法制备,合成方法和条件如下:取原硅酸四乙酯(TEOS)1.50g(国药集团化学试剂有限公司,分析纯),乙醇1.82g(无锡亚盛化工有限公司,分析纯),加入到6g活性炭中搅拌均匀(江苏竹溪活性炭有限公司,20-50目,分析纯),于管式炉350°C焙烧0.5h得到“SiO2-活性炭”混合物.将0.30gCu(NO3)2·3H2O和6.2g Ce(NO3)3·6H2O(国药集团化学试剂有限公司,分析纯)溶于3.8mL去离子水中配成溶液,然后在搅拌下将该溶液加入到上述的“SiO2-活性炭”混合物中,搅拌均匀后于管式炉550°C焙烧3h,即制得CuOCeO2-SiO2催化剂.采用相同的制备方法,将上述体系中一部分Ce(NO3)3·6H2O用La(NO3)3·6H2O、Pr(NO3)3·6H2O或者Nd(NO3)3·6H2O(国药集团化学试剂有限公司,分析纯)替换,使体系中的铈离子与掺杂稀土金属离子的摩尔比为9:1,制备出的催化剂记为CuO-Ce0.9M0.1O2-SiO2(M=La,Pr,Nd).
2.2 催化剂表征
XRD在日本Rigaku公司SarmtLab衍射仪上进行检测,采用CuKα为射线源(λ=0.15406nm),激发电压为40kV,电流100mA,扫描速率20(°)·min-1,扫描范围2θ=10°-80°.比表面积(BET)测试在日本贝尔公司BELSORPII型吸附仪上进行采用N2吸附法进行,样品在200°C下真空预处理3h,然后在-196°C下进行吸脱附实验.TEM是在日本JEM-2100型投射电子显微镜上进行测试的.Raman光谱是在ConfocalRenishaw公司的RM-1000型表面增强激光拉曼光谱仪上进行测试,激光光源采用He-Cd光源器,波长为514nm,扫描范围100-2000cm-1,将在每个样品表面的三个不同位置重复扫描三次后的结果进行重合,对照标准样品的激光拉曼光谱(LRS)谱进行峰的指认.XPS实验使用ThermoESCALAB250型X射线光电子能谱仪测试,实验中使用AlKα光源(1486.6eV),能量分辨率20eV,实验数据以C1s谱的标准结合能位置(284.6eV)为基准校准.H2-TPR实验在美国麦克公司AutoChemⅡ2920上进行,称取50mg样品置于石英反应管中,通入高纯Ar并升温至200°C预处理1h,冷却至50°C,然后再通入H2(10%)-Ar(90%)的还原气,开始程序升温还原至900°C,气体流速为50mL·min-1,升温速率为10°C·min-1,采用热导检测器(TCD)检测.
2.3 催化剂的活性评价
催化剂活性评价是在固定床管式反应器中进行,反应管内径为24mm,热电偶套管外径为4mm,催化剂装填量为5g,粒径为0.45-1.25mm(16-40目),用十倍石英砂对催化剂进行稀释,将其装填于石英反应器中部.反应温度为350-410°C,反应气体为HCl和O2,HCl流量为80mL·min-1,V(O2):V(HCl)=1,反应产物用KI溶液进行吸收后,用碘量法及酸碱滴定法测定生成的氯气及未反应的氯化氢,计算出氯化氢的转化率(XHCl):

3 结果与讨论
3.1 CuO-Ce0.9M0.1O2-SiO2表征
3.1.1 XRD表征
CuO-Ce0.9M0.1O2-SiO2催化剂的XRD谱图如图1所示.可以看到,CuO-CeO2-SiO2的衍射图谱出现对应于CeO2立方萤石结构的特征衍射峰;在15°-20°出现对应于SiO2短程有序、长程无序的无定型形式的特征衍射峰;18未观察到对应于晶相CuO的特征衍射峰,这表明Cu物种可能以氧化物形式在CeO2-SiO2表面,高度分散或者部分可能掺入CeO2的晶格中.从CuO-Ce0.9M0.1O2-SiO2的衍射图谱看到对应于无定型SiO2的特征衍射峰出现偏移,这表明稀土掺杂后导致SiO2短程结构发生了一定的改变.此外,只观察到对应于CeO2立方萤石结构的特征衍射峰,而特征峰均向低角度偏移,这表明掺杂的稀土金属进入了CeO2的晶格中并形成了固溶体结构.16根据Bragg公式计算的各催化剂的晶胞参数列于表1.从表1可以看到,稀土掺杂后的催化剂晶胞参数均变大,这是因为离子半径比Ce4+(0.097nm)更大的Pr3+(0.113nm),Nd3+(0.111nm)和La3+(0.110nm)进入了CeO2的晶格中,导致了晶胞的扩张.此外,由Scherrer公式计算的各催化剂的平均晶粒尺寸列于表1.可以看到,采用模板法制备的CuO-CeO2-SiO2晶粒尺寸为5.1nm,稀土掺杂后可以获得更小的晶粒尺寸.其中,CuO-Ce0.9La0.1O2-SiO2平均晶粒尺寸最小仅为4.1nm.
3.1.2 N2吸附-脱附测试

图1 CuO-Ce0.9M0.1O2-SiO2催化剂的XRD谱图Fig.1 XRD patternsof CuO-Ce0.9M0.1O2-SiO2catalysts
图2是CuO-Ce0.9M0.1O2-SiO2催化剂的N2吸脱附等温线.从图中可以看出催化剂的N2吸脱-脱附等温线属于IV型等温线,表明所有样品均具有介孔结构.此外,H3型回滞环的存在表明这些催化剂的孔道并不规则,它们具有主要为堆积孔构成的混合孔道结构.CuO-Ce0.9M0.1O2-SiO2催化剂的比表面积和孔容数据列于表1中,可见各催化剂孔容比较接近,为0.2157-0.2597m3·g-1.而CuO-Ce0.9M0.1O2-SiO2催化剂均具有较高比表面积,CuO-CeO2-SiO2为127.7 m2·g-1,稀土掺杂后催化剂比表面积有所提高,这主要归因于稀土的掺入固溶体抑制了其个体在焙烧过程中的结晶,19从而导致了较小的晶粒尺寸,显示出更大的比表面积.其中,CuO-Ce0.9La0.1O2-SiO2比表面积显著提高至151.9m2·g-1.

表1 CuO-CeO2-SiO2和CuO-Ce0.9M0.1O2-SiO2催化剂的物理化学性质Table1 Physicalchem icalpropertiesof CuO-CeO2-SiO2and CuO-Ce0.9M0.1O2-SiO2catalysts
3.1.3 TEM结果
CuO-Ce0.9M0.1O2-SiO2催化剂的TEM图见图3.可以看到,CuO-CeO2-SiO2催化剂的晶粒尺寸由6-8nm左右的颗粒不规则排列而成,稀土掺杂后催化剂晶粒尺寸更小,粒径为6nm以下.从图3中选区电子衍射图可以清晰观测到的Pr、Nd和La催化剂的CeO2颗粒的(111)面的晶格条纹,测量的晶面间距为0.320nm,掺杂Pr、Nd和La后(111)面的晶面间距分别增至0.322、0.322和0.326nm,这表明稀土掺杂进入CeO2的晶格并导致了晶胞扩张,这与XRD结果相一致.此外,对CuO-Ce0.9M0.1O2-SiO2催化剂任选区域的元素的含量进行检测,如图4所示.可以看到在不同区域,稀土金属Pr、Nd和La分别在CuOCe0.9La0.1O2-SiO2、CuO-Ce0.9Nd0.1O2-SiO2和CuOCe0.9Pr0.1O2-SiO2催化剂的表面区域的分散是较为均匀的.
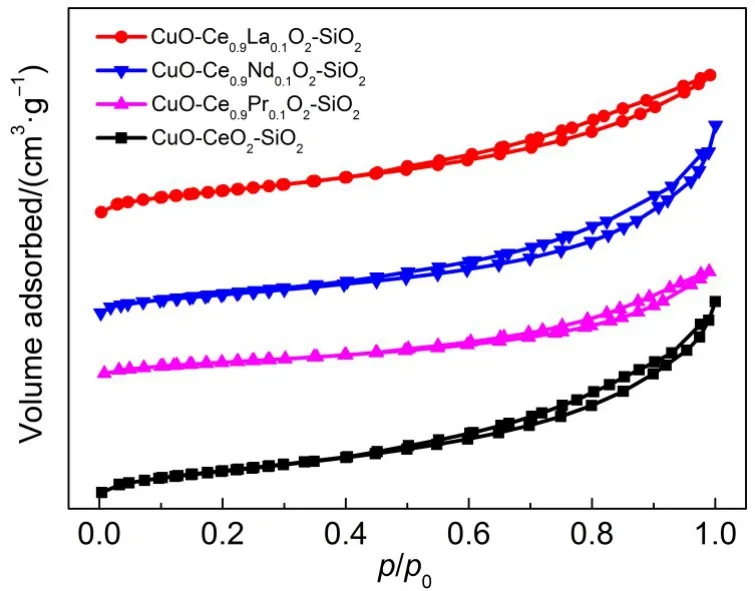
图2 CuO-Ce0.9M0.1O2-SiO2催化剂的N2吸脱附等温线Fig.2 Isotherm of N2adsorption-desorption for CuO-Ce0.9M0.1O2-SiO2catalysts

图3 CuO-Ce0.9M0.1O2-SiO2催化剂的TEM图及选区电子衍射图(插图)Fig.3 TEM imagesand selected area electron diffraction patterns(insets)of CuO-Ce0.9M0.1O2-SiO2catalysts
3.1.4 Ram an表征
CuO-Ce0.9M0.1O2-SiO2催化剂的Raman谱图如图5所示.CuO-CeO2-SiO2在459和605cm-1处只出现了两个吸收峰,459cm-1处的峰归属于CeO2立方萤石结构的F2g对称振动,20而605cm-1处的峰归属为氧空位不对称振动.21掺杂稀土后,对应于CeO2立方萤石结构的F2g对称振动的吸收峰发生移动,这进一步证明了稀土金属掺入CeO2的晶格形成了固溶体结构.在相同检测条件下,催化剂在605cm-1处的吸收峰和在459cm-1处峰振动峰的强度比(A605/A459)与催化剂表面氧空位浓度成正比.22,23根据表1给出CuO-Ce0.9M0.1O2-SiO2催化剂的A605/A459结果,稀土的掺杂可以显著提高固溶体的表面氧空位浓度,CuOCe0.9La0.1O2-SiO2氧空位浓度高达41.43%.
3.1.5 XPS表征
XPS测试能够了解到催化剂表面组成和元素价态等信息,对CuO-CeO2-SiO2样品进行了XPS测试.催化剂表面Ce3d的光电子能谱见图6,Ce3d的光电子能谱出现了八个特征峰.其中u、u''和u'''为Ce4+3d3/2自旋-轨道多重谱线,v、v''和v'''为Ce4+3d5/2自旋-轨道多重谱线,u'和v'则分别为Ce3+3d3/2和Ce3+3d5/2的谱线.Ce3+相对含量可以由v'和u'拟合峰面积与总峰面积估算得到(表1).可以看到,稀土掺杂后催化剂含有更高的Ce3+浓度,而Ce3+浓度的提高直接代表着催化剂表面形成了更多的氧空位和不饱和化学键.其中表面Ce3+浓度顺序为:CuOCe0.9La0.1O2-SiO2>CuO-Ce0.9Nd0.1O2-SiO2>CuO-Ce0.9Pr0.1O2-SiO2>CuO-CeO2-SiO2,这与Raman测得的氧空位浓度顺序是一致的.另外,催化剂的Pr3d、Nd3d和La 3d的XPS图谱如图7所示,Pr3d在933.8和953.0 eV出现对应于Pr3d5/2和Pr3d3/2能级的峰,这表明CuO-Ce0.9Pr0.1O2-SiO2催化剂中Pr元素存在+3和+4多种价态.24CuO-Nd0.9Pr0.1O2-SiO2的Nd3d在983.1 eV出现对应于Nd3d5/2的结合能峰,而CuOCe0.9La0.1O2-SiO2在833.6、838.0、851.1和854.8eV可以看到La3d的裂开的结合能的峰,这与配位基(O 2p)至金属元素(La4f)之间的旋转轨道交互作用以及电子转移有关,25这表明Nd和La主要是以单种氧化价态形式存在.样品的表面铜、稀土元素与表面铈的含量比在表1给出.表面稀土元素与表面铈含量比和投入比例比较接近(10.0%),而各样品中表面铜含量均远大于试剂投入比例(8.7%),显示铜物种在表面富集,表明铜物种主要分散在催化剂的表面.

图4 CuO-Ce0.9La0.1O2-SiO2(a,b),CuO-Ce0.9Nd0.1O2-SiO2(c,d)和CuO-Ce0.9Pr0.1O2-SiO2(e,f)的能量散射X线能谱(EDX)谱图Fig.4 Energy dispersive X-ray spectroscopy(EDX) spectra of CuO-Ce0.9La0.1O2-SiO2(a,b), CuO-Ce0.9Nd0.1O2-SiO2(c,d),and CuO-Ce0.9Pr0.1O2-SiO2(e,f)
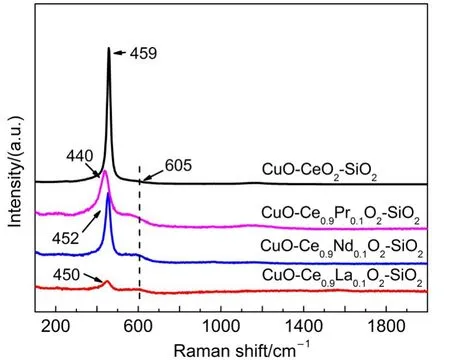
图5 CuO-Ce0.9M0.1O2-SiO2催化剂的Raman图谱Fig.5 Raman spectroscopy of CuO-Ce0.9M0.1O2-SiO2catalysts

图6 CuO-Ce0.9M0.1O2-SiO2催化剂的Ce3d XPS图谱Fig.6 Ce3d XPSpatternsof CuO-Ce0.9M0.1O2-SiO2catalysts
3.1.6 H2-TPR结果
CuO-Ce0.9M0.1O2-SiO2催化剂的H2-TPR谱图如图8所示.CuO-CeO2-SiO2催化剂在230ºC左右出现较强的还原峰,可认为这主要归属于在固溶体表面高分散的CuO的还原,15,26同时也可能含有少量固溶体中Cu2+的还原;27在480和700ºC左右出现两个还原峰,这应归属于CeO2的两步还原:较低温度时发生CeO2表面氧物种的还原以及在较高温CeO2体相氧物种的还原.25CuO-Ce0.9M0.1O2-SiO2催化剂对应于铜物种的还原峰未出现明显的移动,且除CuOCe0.9La0.1O2-SiO2外其还原耗氢量未有明显的提升(表1).但是,归属于CeO2表面氧物种的还原峰强度有所增加,同时归属于CeO2体相氧物种的还原峰均明显向低温移动,这可能与氧空位浓度的提高有关.因为稀土掺入CeO2的晶格中形成了固溶体结构并提高了氧空位浓度,在一定程度上能够促进氧物种在晶格中的流动,这增强了表面氧和体相氧物种之间的平衡,使部分的体相氧物种能够在较低温度时被还原.总而言之,稀土的掺杂未能明显改变铜物种的氧化还原能力,但固溶体的表面氧空位浓度却影响了晶格中氧物种的氧化还原性能.

图7 CuO-Ce0.9M0.1O2-SiO2催化剂的Pr 3d,Nd 3d和La 3d的XPS图谱Fig.7 Pr 3d,Nd 3d,and La 3d XPSpatternsof CuO-Ce0.9M0.1O2-SiO2catalysts

图8 CuO-Ce0.9M0.1O2-SiO2和Ce0.9M0.1O2-SiO2催化剂的H2-TPR图Fig.8 H2-TPR profilesof CuO-Ce0.9M0.1O2-SiO2and Ce0.9M0.1O2-SiO2catalysts

图9 不同反应温度下催化剂的活性Fig.9 Catalytic activitiesof catalystsw ith different reaction tem peratures
3.2 CuO-Ce0.9M0.1O2-SiO2催化剂的性能测试
图9是CuO-Ce0.9M0.1O2-SiO2催化剂在不同温度下活性测试结果.可以看到,CuO-Ce0O2-SiO2拥有良好的催化活性,且随温度的升高显著提高,410ºC时,氯化氢的转化率为55.0%.稀土掺杂后催化剂的活性进一步提高,CuO-Ce0.9La0.1O2-SiO2在410ºC的氯化氢转化率提升至66.8%.各催化剂的活性顺序为:CuO-Ce0.9La0.1O2-SiO2>CuO-Ce0.9Nd0.1O2-SiO2>CuO-Ce0.9Pr0.1O2-SiO2>CuO-CeO2-SiO2.
结合CuO-Ce0.9M0.1O2-SiO2催化剂的表征结果,稀土掺入CeO2晶格中形成了固溶体结构,获得更小的晶粒尺寸和更大的比表面积,并且显著增强了表面氧空位浓度.固溶体的结构(尤其表面氧空位的浓度)显著影响了其氯化氢催化氧化活性.表面氧空位浓度越高的CuO-Ce0.9La0.1O2-SiO2催化剂,其催化活性也越高.因此,可认为固溶体的表面氧空位浓度的提高是导致氯化氢催化氧化性能获得提升的关键.
3.3 Ce0.9M0.1O2-SiO2催化剂表征和性能测试
对Ce0.9M0.1O2-SiO2(M=La,Pr,Nd)催化剂的结构和性能的研究,一定程度上能够有效地验证稀土掺入所形成固溶体结构的差异,是否为各催化剂性能有所不同的关键.Ce0.9M0.1O2-SiO2催化剂的表征结果列于表2.从表2可见,掺杂稀土后,催化剂的晶格常数均变大,这表明稀土进入了CeO2晶格形成了固溶体结构.从Scherrer公式和BET法计算的平均晶粒尺寸和比表面积结果看到,Ce0.9M0.1O2-SiO2可以获得更小的晶粒尺寸和显著增大的比表面积.此外, Raman结果显示催化剂的表面氧空位浓度均显著提高,其中Ce0.9La0.1O2-SiO2约为CeO2-SiO2表面氧空位的3倍.从图8中的Ce0.9M0.1O2-SiO2催化剂的H2-TPR结果看到,在低温出现的两个还原峰(β1、β2)均对应于CeO2表面氧物种的还原,其峰强度明显增大且向高温偏移,而在高温出现对应于CeO2体相氧物种的还原峰(γ),峰强度减小并明显向低温移动.结合Ce0.9M0.1O2-SiO2催化剂的Raman结果,更高的氧空位浓度能够增强表面氧和体相氧物种的之间的平衡,使更多的体相氧物种能够在较低温被还原.对比Ce0.9M0.1O2-SiO2和CuO-Ce0.9M0.1O2-SiO2催化剂的表征结果,稀土掺杂均能形成良好的固溶体结构,获得更小的晶粒尺寸和更大的比表面积,且均不同程度的增强了表面氧空位浓度以及表面氧和体相氧物种之间的平衡.
Ce0.9M0.1O2-SiO2催化剂在不同温度下活性测试如图9所示.CeO2-SiO2具备一定的催化活性,410ºC时氯化氢的转化率为38.5%.稀土掺杂后催化剂的活性却不同程度地降低,各催化剂的活性顺序为: Ce0.9La0.1O2-SiO2<Ce0.9Nd0.1O2-SiO2<Ce0.9Pr0.1O2-SiO2<CeO2-SiO2.对比Ce0.9M0.1O2-SiO2和CuO-Ce0.9M0.1O2-SiO2催化剂的性能结果,稀土掺杂后,CuOCe0.9M0.1O2-SiO2催化活性显著提高,Ce0.9M0.1O2-SiO2催化活性却显著降低.另外,对于CuO-Ce0.9M0.1O2-SiO2,氧空位浓度越高其催化活性越高,而对于Ce0.9M0.1O2-SiO2,较高的氧空位浓度却导致了更低的催化活性.显然,稀土掺杂导致了氧空位浓度的增加以及表面氧和体相氧物种之间的平衡的增强,并不能直接增强固溶体表面发生的氯化氢氧化反应.
3.4 反应级数测试

表2 Ce0.9M0.1O2-SiO2催化剂的物理化学性质Table2 Physicalchem icalpropertiesof Ce0.9M0.1O2-SiO2catalysts

表3 各催化剂对O2和HCl的反应级数Table 3 Reaction ordersof O2and HClof the catalysts
为了更加深入了解稀土掺杂对催化剂的氯化氢氧化表面反应过程的影响,对CuO-CeO2-SiO2, CuO-Ce0.9La0.1O2-SiO2,CeO2-SiO2和Ce0.9La0.1O2-SiO2四个催化剂的反应级数进行了测定.由于HCl催化氧化反应为可逆反应,反应速率的幂函数方程可以表达为:

式中,pO2和pHCl分别为反应物O2和HCl的分压,pCl2和pH2O则分别为逆反应时Cl2和H2O的分压.其中α、β、γ 和δ分别为催化剂对O2、HCl、Cl2和H2O的反应级数.在催化剂动力学测试过程中将转化率限制在5%-20%,故反应式中因此这里忽略逆反应的影响并简化方程如下:

反应级数α,β可采用微分法测得.测试时以N2为平衡气,保持HCl的分压(pHCl)不变,测得不同pO2下rHCl值,以lnpO2对lnrHCl作图,斜率即为α,同理可测得β.
各催化剂对O2和HCl的反应级数测试结果列于表3.可以看到,CeO2-SiO2对O2和HCl的反应级数分别为0.50和0.42,这表明O2分压和HCl分压均显著影响氯化氢氧化反应速率.在反应过程中,生成的Cl2首先会吸附于催化剂的表面,而O2和HCl作为反应物将会与生成的Cl2在催化剂表面产生竞争吸附,因此更高的O2分压以及HCl分压均会加快Cl2的脱附,从而提高Cl2产率.可依此推断,在此过程中Cl2的脱附是这个反应过程的决速步骤.CuOCeO2-SiO2对O2和HCl的反应级数也较大,分别为0.46和0.73,表明Cl2的脱附仍为反应过程的决速步骤.但是CuO-CeO2-SiO2对HCl的反应级数明显更大,此时HCl的分压对氯化氢氧化反应速率的影响更加显著,这主要因为CuO的存在能够提供更多的活性点位用来吸附和活化HCl.
稀土掺杂后,Ce0.9La0.1O2-SiO2和CuO-Ce0.9La0.1O2-SiO2对O2的反应级数分别增大到0.77和0.65,而对HCl的反应级数均显著较小至0.10和0.15,这说明O2分压对氯化氢氧化反应速率的影响远大于HCl分压,此时氧分子的吸附成为这个反应过程的决速步骤.
3.5 机理分析
根据前期的研究工作,15CuO-Ce0.9M0.1O2-SiO2催化剂上氯化氢氧化表面反应过程可用图10表示. HCl主要通过以下三种途径发生氧化反应:(1)这一过程主要遵循Langmuir-Hinshelwood机理,28HCl和O2均吸附于表面高分散的CuO上并发生反应.(2)这一个过程比较复杂,可以用Mar-van-Krevelen机理进行描述:一分子HCl首先与固溶体的晶格氧发生反应生成―OH,Cl原子会占据氧空位,另一分子HCl与―OH反应生成H2O,生成H2O脱附后留下的氧空位被Cl原子占据,占据氧空位的Cl原子活化到固溶体的表面并生成Cl2,而O2是通过氧空位进行吸附和活化.11,29(3)这一过程可以被认为是对途径(1)的补充,O2通过氧空位实现氧的吸附,并存在“氧溢流”现象:氧物种从固溶体的溢流至在其表面高分散CuO上,并与在高分散CuO上吸附的HCl发生反应.对于CuO-CeO2-SiO2催化剂,其表面发生的反应主要是通过途径(1)和(3)上进行的.
对于CeO2-SiO2催化剂,其表面反应过程为途径(2).稀土改性后,氧空位浓度显著增强但催化活性却有所下降.根据密度泛函理论计算研究结果,稀土掺杂后导致固溶体晶胞扩张,使较低的O2分压下(V(O2):V(HCl)=1),占据氧空位的Cl原子更难被O所取代并进行脱附,从而导致了更低的反应速率.17反应级数结果表明CuO-Ce0.9M0.1O2-SiO2与Ce0.9M0.1O2-SiO2催化剂一样,分子氧从催化剂氧空位的的吸附和活化为其决速步骤,此时途径(2)的反应速率同样会降低.然而,此时催化剂表面的反应还存在途径(1)和(3).稀土掺杂增强了固溶体的表面氧空位浓度,使固溶体表面氧空位吸附的氧物种快速溢流至表面高分散CuO上,并与在CuO上吸附的HCl发生反应.可见,稀土掺杂后固溶体表面更高的氧空位浓度增强了固溶体表面的“氧溢流”,使进行途径(3)的比重更大,加快了催化剂氯化氢氧化的整体反应速率,这是CuO-Ce0.9M0.1O2-SiO2具备更高活性的关键.
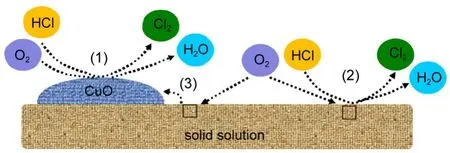
图10 CuO-Ce0.9M0.1O2-SiO2催化剂上氯化氢氧化表面反应过程图Fig.10 Possible reaction pathwaysof HC loxidation on the surface of CuO-Ce0.9M0.1O2-SiO2catalyst
4 结论
采用模板法制备了CuO-CeO2-SiO2以及稀土掺杂的CuO-Ce0.9M0.1O2-SiO2催化剂.表征结果显示稀土掺杂进入CeO2形成良好的固溶体结构,导致更小的晶粒尺寸和更高的比表面积以及显著增强的表面氧空位浓度.性能测试结果显示,稀土掺杂的CuO-Ce0.9M0.1O2-SiO2催化剂的氯化氢氧化反应活性均显著提升,且固溶体氧空位浓度的高低与氯化氢氧化活性直接相关.通过与Ce0.9M0.1O2-SiO2催化剂的结构和性能的对比,发现氧空位浓度的提高不能导致固溶体表面发生的氯化氢氧化反应活性的提升.稀土掺杂后分子氧的吸附成为反应过程的决速步骤,但在V(O2):V(HCl)=1条件下,更高的氧空位浓度反而降低了固溶体表面的反应速率.机理分析认为,对于CuO-Ce0.9M0.1O2-SiO2,稀土掺杂提高了固溶体的表面氧空位,这能够增强固溶体表面的“氧溢流”:氧空位吸附的氧物种快速溢流至表面高分散CuO上,并与在CuO上吸附的HCl发生反应,加快了催化剂氯化氢氧化的整体反应速率.这是CuO-Ce0.9M0.1O2-SiO2具备高活性的关键.
(1)Pérez-Ramírez,J.;Mondelli,C.;Schm idt,T.;Schlüter,O.F.K.; Wolf,A.;M leczko,L.;Dreier,T.Energy Environ.Sci.2011,4, 4786.doi:10.1039/c1ee02190g
(2)Deacon,H.Manufactureof Chlorine.U.S.Pat.85370A,1868.
(3)Crihan,D.;Knapp,M.;Zweidinger,S.;Lundgren,E.; Weststrate,C.J.;Andersen,J.N.;Seitsonen A.P.;Over,H. Angew.Chem.Int.Edit.2008,47,2131.
(4)Tang,J.H.;Chen,X.;Fei,Z.Y.;Zhao,J.H.;Cui,M.F.;Qiao, X.Ind.Eng.Chem.Res.2013,52,11897.doi:10.1021/ ie400200g
(5)Seki,K.Catal.Surv.Asia 2010,14,168.doi:10.1007/s10563-010-9091-7
(6)Mondelli,C.;Am rute,A.P.;Krumeich,F.;Schm idit,T.;Pérez-Ramírez,J.ChemCatChem 2011,3,657.doi:10.1002/ cctc.201000424
(7)Mondelli,C.;Am rute,A.P.;Schmidt,T.;Pérez-Ramírez,J. Chem.Commun.2011,47,7173.
(8)Hernandez,W.Y.;Laguna,O.H.;Centeno,M.A.;Odriozola,J. A.J.Solid State Chem.2011,184,3014.doi:10.1016/j. jssc.2011.09.018
(9)Cao,H.Y.;Wang,J.L.;Yan,S.H.;Liu,Z.M.;Gong,M.C.; Chen,Y.Q.Acta.Phys.-Chim.Sin.2012,28(8),1936.[曹红岩,王健礼,闫生辉,刘志敏,龚茂初,陈耀强.物理化学学报, 2012,28(8),1936.]doi:10.3866/PKU.WHXB201205173
(10)Wang,S.Y.;Li,N.;Luo,L.F.;Huang,W.X.;Pu,Z.Y.;Wang, J.W.;Hu,G.S.;Luo,M.F.;Lu,J.Q.Appl.Catal.B:Environ. 2014,144,325.
(11)Am rute,A.P.;Mondelli,C.;Moser,M.;Novell-Leruth,G.; López,N.;Rosenthal,D.;Farra,R.;Schüster,M.E.;Teschner, D.;Schm idt,T.;Pérez-Ramírez,J.J.Catal.2012,286,287.doi: 10.1016/j.jcat.2011.11.016
(12)Moser,M.;Mondelli,C.;Schm idt,T.;Girgsdies,F.;Schüster, M.E.;Farra,R.;Szentm iklósi,L.;Teschner,D.;Pérez-Ramírez, J.Appl.Catal.B:Environ.2013,132-133,123.
(13)Am rute,A.P.;Larrazábal,G.O.;Mondelli,C.;Pérez Ramírez, J.Angew.Chem.Int.Edit.2013,52,9772.doi:10.1002/ ange.201304254
(14)Chen,X.;Lü,G.M.;Tang,J.H.;Cui,M.F.;Zhou,Z.;Cao,R.; Qiao,X.J.Chem.Eng.Chin.Univ.2011,25,109.[陈 献,吕高明,汤吉海,崔咪芬,周 哲,曹 锐,乔 旭.高校化学工程学报,2011,25,109.]
(15)Fei,Z.Y.;Liu,H.Y.;Dai,Y.;Ji,W.J.;Chen,X.;Tang,J.H.; Cui,M.F.;Qiao,X.Chem.Eng.J.2014,257,273.doi:10.1016/ j.cej.2014.07.033
(16)Jampaiah,D.;Tur,K.M.;Ippolito,S.J.;Sabri,Y.M.;Tardio,J.; Bhargava,S.K.;Reddy,B.M.RSC.Adv.2013,3,12963.doi: 10.1039/c3ra41441h
(17)Farra,R.;García-Melchor,M.;Eichelbaum,M.;Hashagen,M.; Frandsen,W.;A llan,J.;Girgsdies,F.;Szentm iklósi,L.;López, N.;Teschner,D.ACSCatal.2013,3,2256.doi:10.1021/ cs4005002
(18)Jiang,J.T.;Wei,X.J.;Xu,C.Y.;Zhou,Z.X.;Zhen,L.J.Magn. Magn.Mater.2013,334,111.doi:10.1016/j.jmmm.2012.12.036
(19)Hernandez,W.Y.;Laguna,O.H.;Centeno,M.A.;Odriozola,J. A.J.Solid State Chem.2011,184,3014.doi:10.1016/j. jssc.2011.09.018
(20)Si,R.;Zhang,Y.W.;Li,S.J.;Lin,B.X.;Yan,C.H.J.Phys. Chem.B 2004,33,12481.
(21)Meng,Z.H.;Yang,P.;Zhou,R.X.Acta Phys.-Chim.Sin.2013, 29(2),391. [孟中华,杨 鹏,周仁贤.物理化学学报,2013, 29(2),391.]doi:10.3866/PKU.WHXB201212072
(22)Yang,D.;Wang,L.;Sun,Y.Z.;Zhou,K.J.Phys.Chem.C 2010,114,8926.doi:10.1021/jp912227p
(23)Liu,L.;Yao,Z.;Deng,Y.;Gao,F.;Liu,B.;Dong,L. ChemCatChem 2011,3,978.doi:10.1002/cctc.v3.6
(24)Reddy,B.M.;Saikia,P.;Bharali,P.;Park,S.E.;Muhler,M.; Grüunert,W.J.Phys.Chem.C 2009,113,2452.doi:10.1021/ jp809837g
(25)Katta,L.;Sudarsanam,P.;Thrimurthulu,G.;Reddy,B.M.Appl. Catal.B:Environ.2010,101,101.doi:10.1016/j. apcatb.2010.09.012
(26)Gao,X.;Du,X.S.;Cui,L.W.;Fu,Y.C.;Luo,Z.Y.;Cen,K.F. Catal.Commun.2010,12,255.
(27)Menon,U.;Poelman,H.;Bliznuk,V.;Galvita,V.V.;Poelman, D.;Marin,G.B.J.Catal.2012,295,91.doi:10.1016/j. jcat.2012.07.026
(28)Am rute,A.P.;Mondelli,C.;M iguel,A.G.;Hevia,P.J.J.Phys. Chem.C 2011,115,1056.
(29)Farra,R.;W rabetz,S.;Schuster,E.S.;Stotz,E.;Ham ilton,N. G.;Am rute,A.P.;Pérez-Ramírez,J.;López,N.;Teschner,D. Phys.Chem.Chem.Phys.2013,15,3454.doi:10.1039/ c2cp42767b
Effects o f Rare-Earth Add itives on Structures and Perfo rm ances o f CuO-CeO2-SiO2Catalysts fo r Recyc ling Cl2from HClOxidation
XIE Xing-Xing1,2FEIZhao-Yang1,2,*ZOU Chong1,2LIZheng-Zhou1,2CHEN Xian2
TANG Ji-Hai2CUIMi-Fen2QIAO Xu1,2,*
(1State Key Laboratory ofMaterials-Oriented Chemical Engineering,Nanjing Tech University,Nanjing 210009,P.R.China;2College ofChemistry and Chemical Engineering,Nanjing Tech University,Nanjing 210009,P.R.China)
CuO-CeO2-SiO2and rare-earth-doped CuO-Ce0.9M0.1O2-SiO2(M=La,Pr,Nd)catalysts for recycling Cl2from HCloxidation were prepared by a tem platem ethod,using activated carbon as a hard tem p late.The catalyst structures were determ ined using X-ray diffraction(XRD),N2adsorption-desorption,transm ission electronm icroscopy(TEM),Raman spectroscopy,X-ray photoelectron spectroscopy(XPS),and H2temperatureprogrammed reduction(H2-TPR).The catalytic performanceswere also investigated.The results showed thatLa,Pr,and Nd cationswere incorporated into the CeO2lattice and formed nanosized solid solutions;this greatly reduced the catalystgrain sizes,leading to highersurface areas.In addition,the oxygen vacancy concentrations were significantly im proved.The changes in the structures and surface properties of the solid solutions significantly affected the HClcatalytic oxidation performances.The orderof the activities ofvarious catalysts was CuO-Ce0.9La0.1O2-SiO2>CuO-Ce0.9Nd0.1O2-SiO2>CuO-Ce0.9Pr0.1O2-SiO2>CuO-CeO2-SiO2.The oxygen vacancy concentrations of the solid solutions were strongly related to their catalytic activities.However,the structures and performances of the Ce0.9M0.1O2-SiO2catalysts showed thatan increase in the numberofoxygen vacancies resulted in decreased catalytic activities of the solid solutions.Kinetic studies showed thatoxygen adsorption could be the rate-determ ining step for rare-earth-doped catalysts;a higheroxygen vacancy concentration in the solid solution led to a slower reaction rate when the volumetric flow ratio ofO2to HClwas 1.For the CuOCe0.9M0.1O2-SiO2catalysts,spillover of oxygen species in the solid solution into the highly dispersed CuO interfaces was enhanced,which increased the overall reaction rate and gave high activity.
Rare earth;CuO-CeO2;HCl;Catalytic oxidation;Chlorine;Oxygen vacancy
O643
icle]
10.3866/PKU.WHXB201504145 www.whxb.pku.edu.cn
Received:December23,2014;Revised:April14,2015;Published onWeb:April14,2015.
∗Corresponding authors.FEIZhao-Yang,Email:zhaoyangfei@njtech.edu.cn;Tel:+86-25-83587168.QIAO Xu,Email:qct@njtech.edu.cn;
Tel:+86-25-83172298.
The projectwassupported by the National Key Technology Research and DevelopmentProgram of theM inistry of Science and Technology of China (2011BAE18B01),Research and DevelopmentProgram of Jiangsu Province,China(BE2011830),Higher Education Natural Science Foundation of Jiangsu Province,China(13KJB530006),NationalNaturalScience Foundation of China(21306089)and China Postdoctoral Science Foundation (2013M 531340).
国家科技支撑计划(2011BAE18B01),江苏省科技支撑计划(BE2011830),江苏省高校自然科学基金面上项目(13KJB530006),国家自然科学基金(21306089)和中国博士后基金(2013M 531340)资助
©Editorialofficeof Acta Physico-Chim ica Sinica