硫酸根离子对铁在弱碱性溶液中阳极溶解和钝化行为的影响
2015-12-29陈俊劼吕战鹏AHSANEjaz夏小峰刘廷光
陈俊劼 肖 茜 吕战鹏AHSAN Ejaz 夏小峰 刘廷光
(1上海大学材料科学与工程学院材料研究所,上海200072;
2上海大学,省部共建高品质特殊钢冶金与制备国家重点实验室,上海200072)
硫酸根离子对铁在弱碱性溶液中阳极溶解和钝化行为的影响
陈俊劼 肖 茜 吕战鹏*AHSAN Ejaz 夏小峰 刘廷光
(1上海大学材料科学与工程学院材料研究所,上海200072;
2上海大学,省部共建高品质特殊钢冶金与制备国家重点实验室,上海200072)
采用浸泡实验,电化学测试和表面分析技术研究了硫酸根离子浓度对铁在稀碳酸氢钠溶液中开路状态和阳极极化行为的影响.在无硫酸根离子及含有少量硫酸根离子的碳酸氢钠溶液中,铁的开路电位约为(-0.225±0.005)V,并呈现钝化状态,其电化学阻抗很大,腐蚀速率较低.在含有较高浓度硫酸根离子的碳酸氢钠溶液中,铁的开路电位为(-0.790±0.010)V并呈现活性溶解状态,其电化学阻抗较小,腐蚀速率较高,同时阳极极化曲线上能观察到活化-钝化转变现象.由于铁在含有较高浓度硫酸根离子的碳酸氢钠溶液中处于活化状态,阳极极化曲线上存在数个电流峰.足够高的硫酸根离子浓度会导致铁表面预先形成或转变而成的氧化膜失效.相比于自然曝氧状态,在除氧条件下较低的硫酸根离子浓度即可引起铁在碳酸氢钠溶液中由钝态向活性溶解态的转变.
铁;电化学行为;腐蚀失重;钝态;阳极溶解;X射线光电子能谱;电位-pH图
KeyWords:Iron;Electrochem icalbehavior;Corrosion weightloss;Passivity;Anodic dissolution; X-ray photoelectron spectroscopy;Potential-pH diagram
1 Introduction
Bicarbonate and sulfate ions are often present in natural and industrialenvironments.1-21Theelectrochemicalbehaviorof iron, carbon steelsand low alloy steels in bicarbonate-bearing solutions hasbeen studied due to its theoretical importance in understanding of anodic reaction mechanisms,1-15and due to the practical applications such as corrosion in oil and gas fields,corrosion and stress corrosion cracking of gas transmission pipeline steels, corrosion in concrete pore environments,and corrosion in simulating groundwater environments of candidate waste disposal sites.22-36Bicarbonate ionshavemultiple effectson anodic reactions,such as participating anodic dissolution through active adsorption,inhibiting further anodic dissolution by form ing protective film,and enhancing anodic dissolution through dissolving the protective film.Depending on the initial condition, iron in bicarbonate solutions could exhibit two kinds of open circuitstates,passive stateoractive state.5,15Open circuitstatehas a criticaleffecton corrosion kinetics.Open circuitstate could be affected by both passivating species and aggressive species.The effectsof sulfate on corrosion and electrochemicalbehaviorsof iron and steelshave been reported.16-21
Thiswork is designed to investigate if sulfate as an aggressive type anion can affect the type of open circuit states and the resultantanodic dissolution/passivation kinetics.The result could provide a direct judgment on the corrosivity of environments, which is also expected to provide relevant information on the anodic behaviorat the stress corrosion crack tip where aggressive ions could accumulate.The changes of open circuit states and anodic processes of ironw ith sulfate concentration in dilute bicarbonate solutionswere investigated bymeasurements of open circuit potential(OCP),immersion corrosion tests,surface observationsand surface film analysis technology,potentiodynamic polarization measurements and electrochem ical impedance spectroscopymeasurements.The effects of anions on electrode kinetics and thermodynam ics are discussed in terms of potential (E)-pH diagrams and electrochemicalmeasurements.Both aerated and deaerated solutions were used for various filed applicationsand theoreticalanalyses.
2 Experim en tal
Specimenswere fabricated from iron rodswith purity of 99.9%. The electrode sizewas10mm×10mm×8mm.Copperw irewas welded to the back of the iron specimen.The iron electrodewas embeddedw ith epoxy resinwith an exposed areaof 1.0 cm2.The surface of the iron electrodewas grinded to 1200-gritSiC paper then rinsed in ethanol and acetone prior to the electrochem ical measurements.All testswere carried out in naturally aerated and deaerated x mol·L-1NaHCO3+y mol·L-1Na2SO4solutionsat25°C, w ith x of0.02 and 0.05 and y of 0,0.003,0.5,1.All testsolutions were prepared by deionized water and analytical reagents.Deaerationwasachieved by bubbling N2gas into the testsolution for 1 h before themeasurements and N2gas was kept continuous bubbling during the processof experiment.The conductivity and pH valuesof testsolutionsweremeasured.
A three-electrode cellwas used for electrochem icalmeasurements.Open circuit potential,anodic polarization curve,and electrochemical impedance spectroscopy(EIS)weremeasured by using a German Zahner-Zennium electrochemicalworkstation. Theworking electrodewas the pure iron specimen,the counter electrodewas a platinum plate,and the reference electrodewas a saturated calomelelectrode(SCE).A ll potentialsweremeasured and quoted againstSCE if notspeciallymentioned.
Prior to anodic polarization tests,theworking electrodewas immersed in the testsolution for600 sand OCPvalueswere recorded.Anodic polarization curvesweremeasured ata sweep rate of 0.333mV·s-1.Prior to EIS tests,theworking electrodewas immersed in the test solution for 2 h.EISmeasurementswere performed at open circuit potentials.The frequency range was from 100 kHz to 10mHz.The amplitude of sinusoidalexcitation voltagewas 5mV.The data collected was interpreted w ith software Zsimpw in.Each testwas repeated three timesand showed good reproducibility.No prior cathodicalpolarizationwasapplied before OCP test.
Immersion testswere performed using 15mm×15mm×3mm coupons.The couponswere polished to 1200-gritSiC paper,and then rinsed w ith ethanoland acetone in sequencebefore immersion tests.The surface areaswere calculated and,the difference between the initial weights and the final weight(ΔW),was measured.Average corrosion rates(vcorr)inmm·year-1were calculated.The surfacemorphologiesof specimensw ith corrosion products removed after immersion tests were observed w ith CamScan Apollo 300 thermal field em ission scanning electron m icroscope(SEM).
The samples for X-ray photoelectron spectroscopy(XPS)tests were5mm×5mm×3mm iron coupons immersed in testsolutions for 2 h.XPSanalysiswas performed by using a Japan Physical Electronics Quantum 2000 Scanning ESCA M icroprobe.Spectra were generated using a focused monochromatic A l KαX-ray source operated at25.1W(1486.6 eV)and passenergy of 58.7 eV.The ion gunwasoperatedw ith an Ar+atenergy of 2 kV and the sputtering beam currentwas29 nAwith an incidentangle of 45°.The sputter ratesof 0.25 nm per cycle for first20 cyclesand then 2.5 nm per cyclewere used.Sputter rateswere determ ined by sputtering a known thickness of SiO2on Si using the same sputter conditions.The sputter rateswere for SiO2.The samples measured in an“as received”condition w ith no other surface cleaning treatment.Binding energy calibration was done by linking the reference to C 1s line,thebinding energy of C―C or C―H located at284.8 eV.

Fig.1 Open circuit potential vs imm ersion tim e curves for iron in aerated 0.02mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,0.5,1)solutions

Fig.2 Open circuit potential vs imm ersion tim e curves for iron in aerated 0.05mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,0.5,1)solutions

Fig.3 Open circuit potential vs immersion time curves for iron in deaerated 0.02m ol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,1)solutions
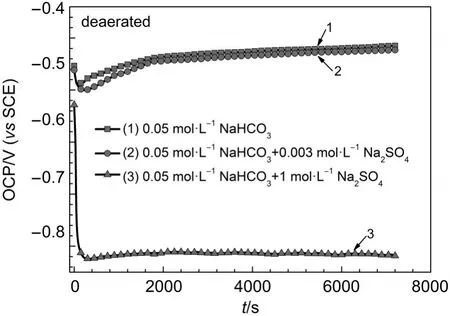
Fig.4 Open circuit potential vs imm ersion time curves for iron in deaerated 0.05m ol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,1)solutions
3 Resu lts
3.1 Open circuit po ten tials
3.1.1 Open circuitpotentials in aerated solutions
Open circuit potential vs immersion time curves for iron in aerated x mol·L-1NaHCO3+y mol·L-1Na2SO4(x=0.02,0.05;y= 0,0.003,0.5,1)solutions are shown in Fig.1 and Fig.2.Open circuit potentials for iron in aerated NaHCO3solution w ithout sulfate orw ith 0.003mol·L-1Na2SO4exhibited a trend ofmoving in the positive direction w ith increasing immersion time.Open circuitpotential for iron in aerated NaHCO3solutionwith 0.5 and 1mol·L-1Na2SO4moved in thenegativedirectionwith increasing immersion time.The initialOCPvalues,OCPt=0h,for iron in 0.02 and 0.05 mol·L-1NaHCO3solutions decreased gradually w ith increasing sulfate concentration from 0 to 1mol·L-1.For iron in NaHCO3solution withoutsulfate orw ith 0.003mol·L-1Na2SO4, OCPvaluesatan immersion time of 2 h,OCPt=2h,were between -0.23 and-0.22V.The differencesbetween OCPt=0hand OCPt=2hwere less than 0.06V,and the electrode surfaceswere brightafter the immersion test.For iron in NaHCO3solution w ith 0.5 and 1 mol·L-1Na2SO4solutions,valuesof OCPt=2hwere in the rangeof -0.80 to-0.78V.The differencesbetween OCPt=0hand OCPt=2hweremore than 0.30V,and the electrode surfacesafter immersion were dark.These results show thatadding sulfate in bicarbonate solutions caused a negative shiftof OCP.OCPvalues for iron in 1mol·L-1Na2SO4with adding 0.02 or0.05mol·L-1NaHCO3were significantly lower than that for iron in 1mol·L-1Na2SO4solution.
3.1.2 Open circuitpotentials in deaerated solutions
Open circuit potential vs immersion time curves for iron in deaerated x mol·L-1NaHCO3+y mol·L-1Na2SO4(x=0.02,0.05;y= 0,0.003,1)solutionsare shown in Fig.3 and Fig.4.Open circuit potentials for iron in deaerated 0.02,0.05mol·L-1NaHCO3and 0.05mol·L-1NaHCO3+0.003mol·L-1Na2SO4solutions firstly decreased and then increased w ith increasing immersion time. Open circuitpotential for iron in deaerated 0.02mol·L-1NaHCO3+ 0.003mol·L-1Na2SO4,0.02mol·L-1NaHCO3+1mol·L-1Na2SO4, and 0.05mol·L-1NaHCO3+1mol·L-1Na2SO4solutionsmoved in the negative directionw ith increasing immersion time.For iron in deaerated 0.02,0.05·L-1NaHCO3and 0.05mol·L-1NaHCO3+ 0.003mol·L-1Na2SO4solutions,valuesof OCPt=2hwerebetween -0.44 and-0.41 V.The differences between OCPt=0hand OCPt=2hwere less than 0.04 V.For iron in deaerated 0.02 mol·L-1NaHCO3+0.003mol·L-1Na2SO4,0.02mol·L-1NaHCO3+1mol·L-1Na2SO4,and 0.05mol·L-1NaHCO3+1mol·L-1Na2SO4solutions,values of OCPt=2 hwere around-0.82 V.The differences between OCPt=0 hand OCPt=2 hweremore than 0.25V.These results are different from those in aerated solutions.Open circuit potentials for passivity state and active dissolution state in deaerated solutions are lower than their corresponding values in aerated solutions.In deaerated 0.02mol·L-1NaHCO3solution,adding 0.003mol·L-1Na2SO4could cause the transition from passivity state to active dissolution state.
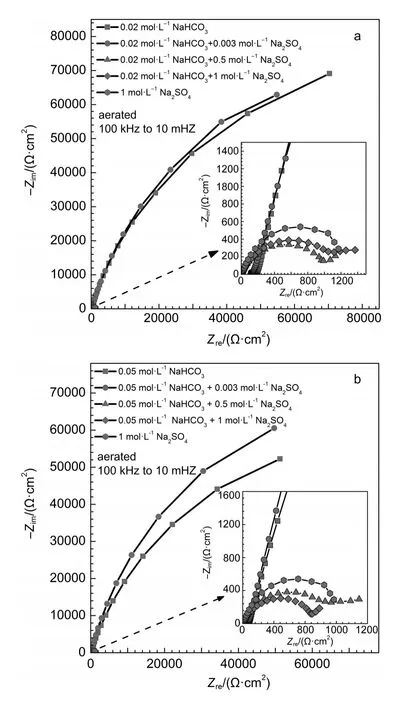
Fig.5 Nyquist im pedance representation plots for iron in aerated (a)0.02m ol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,0.5,1) solutionsand(b)0.05mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0, 0.003,0.5,1)solutions
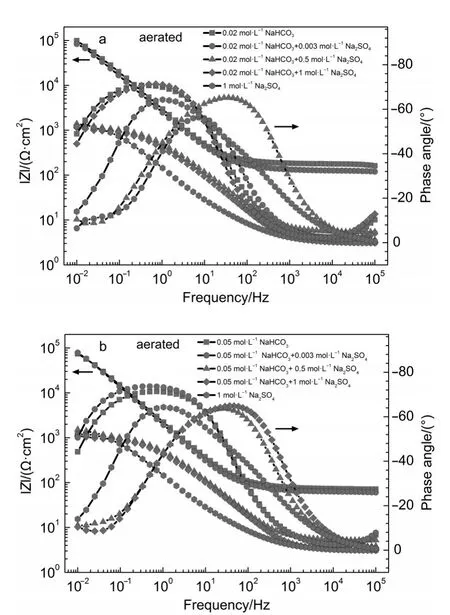
Fig.6 Impedancem odu le rep resentation plots for iron in aerated (a)0.02mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,0.5,1) solutionsand(b)0.05mol·L-1NaHCO3+y m ol·L-1Na2SO4(y=0,0.003,0.5,1)solutions
3.2 Elec trochem ical im pedance spec troscopy
3.2.1 In aerated solutions
EISdiagrams for iron in aerated 1mol·L-1Na2SO4and x mol·L-1NaHCO3+y mol·L-1Na2SO4(x=0.02,0.05;y=0,0.003,0.5,1) solutions are shown in Fig.5 and Fig.6.Based on the reported analysesof similar testsystems,14,33the obtained EIS resultswere fitted and analyzed.For iron in aerated NaHCO3solution w ithout sulfateorw ith 0.003mol·L-1Na2SO4,EIS resultswere fitted with an equivalent circuit for passivity,as shown in Fig.7a.In this equivalent circuit,Rs,Rf,Cf,Cdl,and Rctrepresentsolution resistance,resistanceacross the passive film,passive film capacitance, interfacial capacitance,and charge transfer resistance.The results are summarized in Table 1.For iron in aerated NaHCO3solution w ith 0.5 and 1mol·L-1Na2SO4,EIS resultswere fitted w ith an equivalent circuit for active dissolution,as shown in Fig.7b.In this equivalent circuit,Rs,Cdl,Rctrepresent solution resistance, interfacial capacitance,charge transfer resistance.The results are summarized in Table 2.
Solution resistance Rsdecreased w ith the increase of Na2SO4concentration,show ing the increase of the electrical conductivity of solutions,asshown in Tables1 and 2.Thevaluesof Rctfor iron in aerated NaHCO3solutionw ithoutsulfateorw ith 0.003mol·L-1Na2SO4weremore than 100 kΩ·cm2,show ing that the surface layer formed on theelectrode surfacehad agood protectiveeffect on thematrix.In aerated NaHCO3solutionwith 0.5 and 1mol·L-1Na2SO4,the values of Rctwere far less than those in aerated NaHCO3solution w ithoutsulfate orw ith 0.003mol·L-1Na2SO4, show ing that corrosion of iron would take place under active dissolution.
3.2.2 In deaerated solutions
EISdiagrams for iron in deaerated x mol·L-1NaHCO3+y mol·L-1Na2SO4(x=0.02,0.05;y=0,0.003,1)solutions at25°C are shown in Figs.8 and 9.For iron in deaerated 0.02mol·L-1NaHCO3solution and 0.05mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003)solution,EIS resultswere fitted w ith an equivalent circuit for passivity,as shown in Fig.7a.The resultsare summarized in Table 3.For iron in deaerated 0.02mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0.003,1)and 0.05mol·L-1NaHCO3+1mol· L-1Na2SO4solutions,EIS resultswere fitted w ith an equivalent circuit for active dissolution,as shown in Fig.7b.The results are summarized in Table4.

Fig.7 Equivalent circuitsp roposed for analyzing the electrochem ical im pedance response for iron in x mol·L-1NaHCO3+y m ol·L-1Na2SO4solutions for(a)passive state and(b)active dissolution state

Tab le1 EIS component values for iron in aerated x m ol·L-1NaHCO3+y m ol·L-1Na2SO4(x=0.02,0.05;y=0,0.003)solutions*
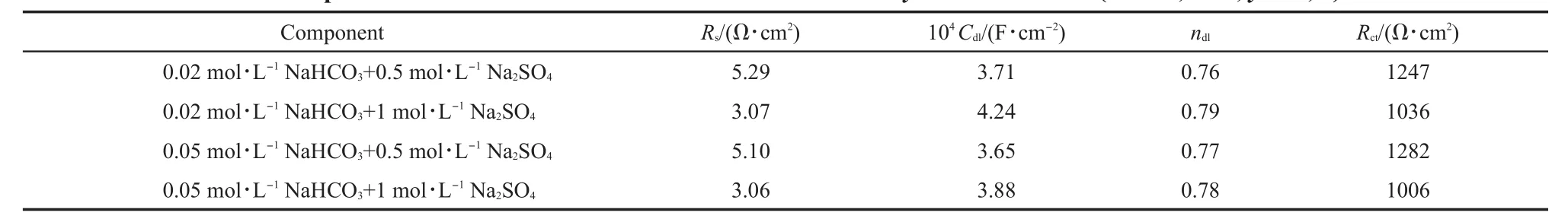
Table2 EIS com ponent values for iron in aerated x m ol·L-1NaHCO3+y m ol·L-1Na2SO4(x=0.02,0.05;y=0.5,1)solutions*
Similarly,solution resistance Rsdecreased with the increase of Na2SO4concentration,as shown in Tables3 and 4.The valuesof Rctfor iron in deaerated 0.05mol·L-1NaHCO3solution w ithout sulfate orw ith 0.003mol·L-1Na2SO4were about100 kΩ·cm2, show ing the presence of a good protective surface layer.In deaerated 0.02mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0.003,1) and 0.05 mol·L-1NaHCO3+1 mol·L-1Na2SO4solutions,the significantly low valuesof Rctindicated the active dissolution of iron.
The valuesof Rctfor iron in deaerated solutionsunder passivity statewere lower than those in aerated solutions.The valuesof Rctfor iron in deaerated solutions underactive dissolution statewere higher than those in aerated solutions.
3.3 Anod ic po larization cu rves
3.3.1 In aerated solutions
Anodic polarization curves for iron in aerated x mol·L-1NaHCO3+y mol·L-1Na2SO4(x=0.02,0.05;y=0,0.003,0.5,1) solutionsat25°C are shown in Fig.10 and Fig.11.Themaximum current imaxoccurred at the potential Emax.Epassive,i,Epassive,f,and Epassiverepresent the initialpotential,finalpotential,and thewidth of the passive region,respectively.ipassiveis the current in the passive region.Increasing the sulfate concentration had negligibleeffects on the Epassive,f.In NaHCO3solutionw ithoutsulfate orw ith 0.003 mol·L-1Na2SO4,no current peak existed on the anodic polarization curvesand no active-passivation transition phenomenon could be found.Adding 0.003mol·L-1Na2SO4slightly increased ipassive.In NaHCO3solutionw ith 0.5 and 1mol·L-1Na2SO4,activepassivation transition appeared on curvesand single ormultiple current peakswere observed.imaxincreased w ith increasing the sulfate concentration.Emax,Epasssive,i,and Epassivefor y=0.5were close to those for y=1.
3.3.2 In deaerated solutions
Anodic polarization curves for iron in deaerated 0.02mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,1)solutionsat25°C are shown in Fig.12.Epassive,fwasnotsensitive to sulfate concentration, while Epassivedecreased w ith increasing sulfate concentration.No currentpeak or active-passivation transition phenomenon could be found on the anodic polarization curves for iron in deaerated 0.02mol·L-1NaHCO3solution.For y=0.003,1,active-passivation transition was observed on polarization curves and multiple currentpeaksappeared due to transition of open circuitstate from passivity to active dissolution state in deaerated solution.ipassivefor iron in deaerated 0.02mol·L-1NaHCO3+1mol·L-1Na2SO4solution was a little higher than that in deaerated 0.02 mol·L-1NaHCO3+0.003mol·L-1Na2SO4solution.imaxfor iron in deaerated 0.02mol·L-1NaHCO3+0.003mol·L-1Na2SO4solutionwas lower than that in deaerated 0.02mol·L-1NaHCO3+1mol·L-1Na2SO4solution.

Fig.8 Nyquist im pedance representation p lots for iron in deaerated(a)0.02mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,1)solutionsand(b)0.05m ol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,1)solutions

Fig.9 Im pedancemodule representation p lots for iron in deaerated(a)0.02m ol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,1)solutionsand(b)0.05mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,1)solutions

Table 3 EIS com ponent values for iron in deaerated x m ol·L-1NaHCO3+y mol·L-1Na2SO4(x=0.02,0.05;y=0,0.003)solutions*
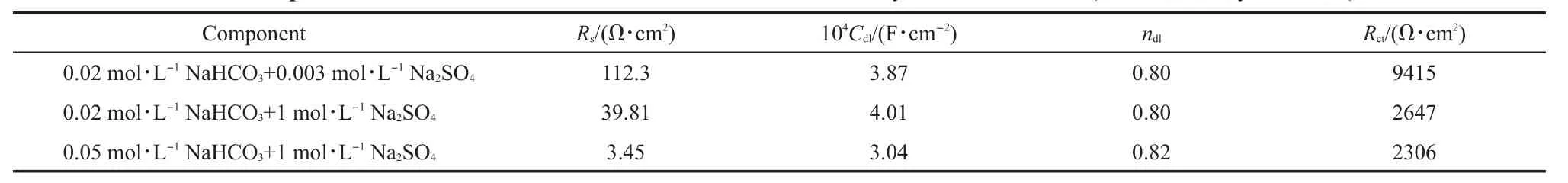
Table4 EIS com ponent values for iron in deaerated x m ol·L-1NaHCO3+y m ol·L-1Na2SO4(x=0.02,0.05;y=0.003,1)solutions*
Anodic polarization curves for iron in deaerated 0.05mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,1)solutionsat25°C are shown in Fig.13.Neither currentpeak nor dissolution/passivation transition could be observed on the anodic polarization curves for y=0 and y=0.003,which was sim ilar to that for iron in aerated solutions.For y=1,several currentpeaksappeared on the anodic polarization curve.
Characteristic parameters of anodic polarization curves in aerated and deaerated solutionsaredifferent.In deaerated 0.02mol· L-1NaHCO3,ipassivewas close to that in aerated 0.02mol·L-1NaHCO3solution.In deaerated 0.02mol·L-1NaHCO3+1mol·L-1Na2SO4,ipassivewashigher than that in aerated solution.Emaxand imaxin deaerated solutionswere lower than those in aerated solutions.
In deaerated 0.05mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0, 0.003)solutions,ipassivevaluesof anodic polarization curveswere also close to those in aerated solutions.In deaerated 0.05mol·L-1NaHCO3+1mol·L-1Na2SO4solution,ipassive,imax,and Emaxwere higher than those in aerated solutions.

Fig.10 Anodic polarization curves for iron in aerated 0.02 m ol·L-1NaHCO3+y m ol·L-1Na2SO4(y=0,0.003,0.5,1)solutions

Fig.11 Anodic polarization curves for iron in aerated 0.05 mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,0.5,1)solutions
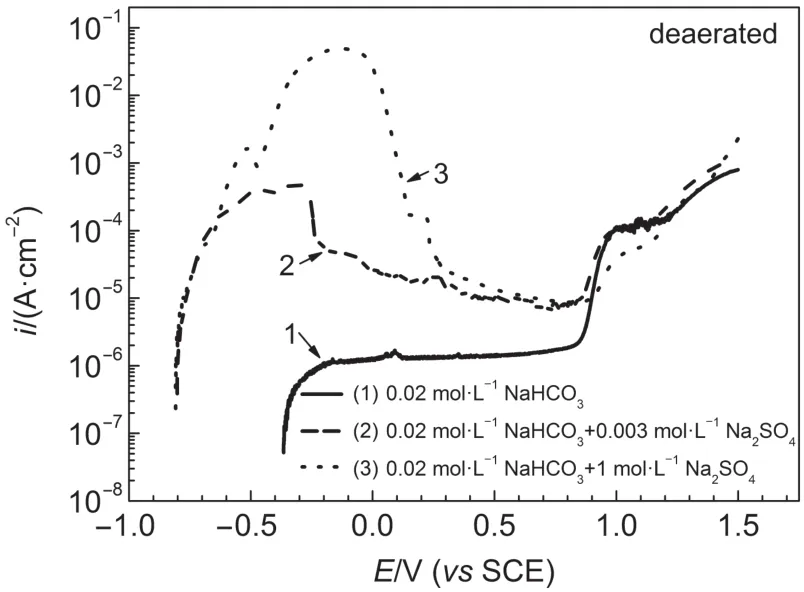
Fig.12 Anodic polarization curves for iron in deaerated 0.02mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,1) solutionsat 25°C

Fig.13 Anodic polarization curves for iron in deaerated 0.05m ol· L-1NaHCO3+y mol·L-1Na2SO4(y=0-1)solutionsat25°C

Fig.14 Weightloss vs test tim e for iron in aerated 0.05mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0.003,1)solutions
3.4 Imm ersion test resu lts
Results from the immersion tests are shown in Fig.14. Weightloss vs immersion time curve for iron in aerated 0.05mol· L-1NaHCO3+1mol·L-1Na2SO4solution exhibited an approximately linear relationship w ithin the test period of 72 h test, leading to a corrosion rateof about0.103mm·year-1atOCPof about-0.80 V.Iron showed a rather smallamountofweightloss fora 72 h immersion test in aerated 0.05mol·L-1NaHCO3+0.003 mol·L-1Na2SO4solution,leading to a corrosion rateofabout5.46× 10-4mm·year-1atOCPofabout-0.30V.Corrosion rate isstrongly affected by sulfate concentration.
After immersion tests in aerated 0.05mol·L-1NaHCO3+y mol· L-1Na2SO4(y=0.003,1)solutions,the corrosion products on the specimen surfaceswere cleanedwith a soft rubberand then observed w ith SEM.The surfacemorphologiesare shown in Fig.15.The specimen surfacesof iron after immersion in aerated 0.05mol·L-1NaHCO3+1 mol·L-1Na2SO4solution were dark and rough, show ing thatactive corrosion occurred during the immersion test. With increasing of immersion time,pitsbecamemore and larger, and grindingmarksbecame less clear,as shown in Fig.15(a-c). The specimens surface of iron was bright after immersion in aerated 0.05mol·L-1NaHCO3+0.003mol·L-1Na2SO4solution and grinding marks during specimen preparation were still visible, show ing that the surfaceswere notsignificantly affected by the immersion procedure,asshown in Fig.15d.
3.5 XPS analysis
The variation in chemical compositionwith XPSdepth profiles of iron,oxygen,and carbon obtained from the XPS survey scans for iron specimens immersed in aerated 0.02mol·L-1NaHCO3, 0.05mol·L-1NaHCO3,and 0.05mol·L-1NaHCO3+0.003mol·L-1Na2SO4solutionsareshown in Fig.16.Asobserved in Fig.16,iron concentration increased w ith depth and oxygen concentration decreased w ith depth as expected.The high carbon concentrationat the surface of each specimenmay be due to organic carbon contam ination during the specimen preparation and postimmersion storage or the formation of FeCO3.The carbon concentration decreasedmarkedly after first sputtering the surface and remained nearly constantafterwards for all specimens.According to Fig.16,oxygen concentrationsdecreased to half of their initialvaluesat the depth of about3 nm.The placewhere oxygen concentration decreases to half of its initial value is the interface of oxideand transition layer.If the distance between the surface and the interface is defined as the thickness of oxide layer,the thicknessof oxide layeron testspecimens isabout3 nm.
Fig.17 and Fig.18 present the high resolution of XPSspectra of iron in 0.05mol·L-1NaHCO3+0.003mol·L-1Na2SO4solution for the Fe 2p3/2,O 1s,and C 1s regionw ith differentsputter depths. Thebinding energies for the peakshavebeen referenced to C―C bond at284.8 eV.As shown in Fig.17,at the sputter depth of 0 nm,FeCO3,Fe2O3,and FeO existed in the surface film.Asshowed in Fig.18,at the sputter depth of 0.9 nm,FeCO3disappeared and Fe2O3and FeOwere stillpresent.Metallic ironwas found atboth depthsbecause the film wasvery thin.Thebinding energy values for fitting are identicalw ith those reported in literature.38,46-50The emergence of FeOmay be due to the sputtering process.46,47,50

Fig.16 XPSdepth profiles for iron,oxygen,and carbon recorded for the iron specim ens imm ersed in aerated(a)0.02m ol·L-1NaHCO3,(b)0.05m ol·L-1NaHCO3,and(c)0.05m ol·L-1NaHCO3+ 0.003mol·L-1Na2SO4solutions
4 Discussion
Previous research showed that the value of pH was approximately 8.4 for the concentrations of bicarbonate solutions from 0.01 to 1.0mol·L-1.3Themeasured conductivity and pH valuesof x mol·L-1NaHCO3+y mol·L-1Na2SO4solutions(x=0.02,0.05;y= 0,0.003,0.5,1)are shown in Fig.19.With the increase of sulfate concentration,the conductivity of x mol·L-1NaHCO3+y mol·L-1Na2SO4solutions(x=0.02,0.05;y=0,0.003,0.5,1)increased.The values of conductivity of 0.05 mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,0.5,1)solutionswerehigher than thoseof0.02 mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,0.5,1)solutions for the same sulfate concentration.The solution pH valuesof0.02 or0.05mol·L-1NaHCO3solutions slightly increasedwith adding 0.003 mol·L-1Na2SO4.The pH value decreased w ith sulfate concentration for y≥0.003.The pH values of 0.05 mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,0.5,1)werehigher than thoseof0.02mol·L-1NaHCO3+y mol·L-1Na2SO4(y=0,0.003,0.5, 1)except for y=1.
According to the thermodynam ic equilibrium calculations,the main ionic species in x mol·L-1NaHCO3+y mol·L-1Na2SO4solutions in thisstudy are Na+,OH-,The anodic behaviorsof iron aremainly the resultsof the combined actionsof anionsOH-,In the dilute solutionsused here, the activity coefficient of HCO3-is 0.863 in a 0.02mol·L-1NaHCO3solution and 0.804 in a 0.05mol·L-1NaHCO3solution.51Thus the activity of HCO3-is 0.017 in a 0.02mol·L-1NaHCO3solution and 0.04 in a0.05mol·L-1NaHCO3solution.PotentialpH diagram of Fe-H2O system isused to analyze the thermodynam ic information of iron in dilute NaHCO3solutions w ith or w ithoutsulfateaddition3,52assum ing[Fe2+]=[Fe3+]=1×10-6mol·L-1. Candidateanodic reactionsare:15,37,40,41


The dominant anodic reactions are dependent on the concentrationsof active speciesand the electrode potential.

Fig.17 High resolution of XPSspectra for iron imm ersed in aerated 0.05m ol·L-1NaHCO3+0.003m ol·L-1Na2SO4solution at the sputter depth of 0 nm

Fig.18 High resolution of XPSspectra for iron immersed in aerated 0.05mol·L-1NaHCO3+0.003m ol·L-1Na2SO4solution at the sputter depth of0.9 nm
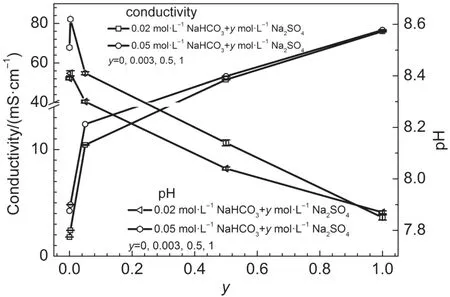
Fig.19 Conductivity and pH valuesof x mol·L-1NaHCO3+y mol· L-1Na2SO4solutions(x=0.02,0.05;y=0,0.003,0.5,1)
In aerated NaHCO3solutionw ithoutsulfateorwith 0.003mol· L-1Na2SO4,the stableOCPvaluesare(-0.225±0.005)or(0.015± 0.005)V(vs SHE),as shown in Fig.1 and Fig.2.These OCP valuesare located in the region of Fe2O3in E-pH diagram of Fe-H2O system,asshown in Fig.20.From the immersion test results,the surfaces of specimens were still bright and grindingmarks were visible,and therewas very low corrosion weight loss after immersing in aerated 0.05mol·L-1NaHCO3+0.003mol·L-1Na2SO4for 72 h,as shown in Fig.14 and Fig.15d.High valuesof Rctfrom the fitting results of EIS data of electrode in abovementioned solutions are shown in Table 1.All these results indicate the high protective property of the surface layer formed on electrode surface.This kind passivity of ironwasmainly caused by the pre-formed oxide film in air.Aftera shortperiod of prior cathoidc polarization,iron could notmaintain the passivity in bicarbonate solutionsof 0.01-1.0mol·L-1.5,15,53Mao etal.31pretreated the X80 pipeline steel in deaerated bicarbonate solutions at-0.95V for30min to reduceair-formed oxide filmsbefore the potentiostatic test.Passive film could not form on bare surface iron in bicarbonate solutions.XPSdata also showed that the inner surface film formed on iron in bicarbonate solutionswere ferric oxide,Fig.18.FeCO3observed only on the outmost layer was thought to the productof the prior formed oxidewith bicarbonate ionsafter immersion in bicarbonate solutions,Fig.17.

Fig.20 Potential-pH(E-pH)diagram of Fe-H2O system considering bicarbonate speciesat 25°C
For iron in aerated NaHCO3solution w ith 0.5 and 1mol·L-1Na2SO4,the stable OCPvaluesare(-0.790±0.010)and(-0.550± 0.010)V(vs SHE),asshown in Fig.3 and Fig.4.According to Eqs. (2)and(3),40theequilibrium potentials for Fe/FeCO3and FeCO3/ Fe2O3are calculated w ith themeasured solution pH values and valuesof HCO3-activity by follow ing equations:
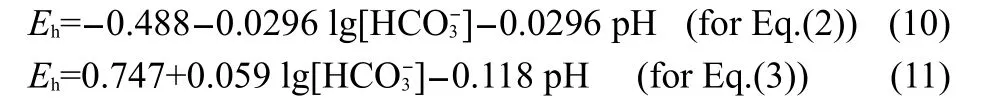
The results are shown in Fig.20.The OCP values for iron in aerated x mol·L-1NaHCO3+y mol·L-1Na2SO4(x=0.02,0.05;y= 0.5,1)solutions are both in the thermodynam ics stability region of FeCO3and Fe2+in E-pH diagram of Fe-H2O system.However, a low reaction resistanceand ahigh corrosion ratewereobserved for iron in aerated 0.05mol·L-1NaHCO3+1mol·L-1Na2SO4solution,as shown in Fig.14,Fig.15b,and Table 2.It is proposed that sulfate ions of sufficiently high concentrations in solutions could degrade the prior formed oxide layeron iron or the transformed oxide layer in bicarbonate solutions.As the result,high concentrations of sulfate anions cause a significantly negative shiftof open circuitpotential.In the absence of protective film, anodic dissolution reactionsof iron or formation reactionsofnonprotective oxide filmsare favored.
The open circuit potential for iron in 1mol·L-1Na2SO4was more positive than those in 1mol·L-1Na2SO4with a low concentration of bicarbonate,show ing thatbicarbonatewould have astrong effecton theelectrified lay at themetal-solution interface.
The open circuit states of iron in deaerated solutions are somewhat different from those in aerated solutions.The open circuit states of iron in aerated and deaerated 0.02 mol·L-1NaHCO3and 0.05mol·L-1NaHCO3solutions are in passivity state.The open circuit states of iron in deaerated 0.02mol·L-1NaHCO3+1mol·L-1Na2SO4and 0.05mol·L-1NaHCO3+1mol· L-1Na2SO4solutions are in active dissolution state.The open circuitstateof iron in deaerated 0.02mol·L-1NaHCO3+0.003mol· L-1Na2SO4solution is in active dissolution state,which is in passive state in the aerated solution.In deaerated solutions,oxygen reduction reaction becomes negligible.Open circuit potentials in deaerated solutions are lower than those in aerated solutions.In aerated solutions,oxygen would facilitate the formation of oxide film or the film transition to form amore protective layer that could notbe attacked by a low concentration sulfate.
Fig.10-Fig.13 show that relatively high concentration sulfate ionshave a significanteffecton anodic polarization behavior.In aerated bicarbonate solutionsw ithoutsulfate,therewasno activepassivation phenomenon on anodic polarization curves.Adding 0.003mol·L-1sulfate in aerated NaHCO3solutionsdoesnothave significant effect on the configuration of anodic polarization curves.Adding 0.5 or1mol·L-1sulfate could causea transition ofopen circuitstate from passivity to active dissolution and activepassive transition in the anodic polarization curves.Similareffects of sulfate on anodic polarization behavior in deaerated bicarbonate solutionsare observed,buta lower concentration of sulfate isnecessary to attack the passive film.
A high concentration of sulfate would result in a high conductivity and facilitate the dissolution of surface film,which thus would hinder the formation of protective film on iron in bicar-bonate solutions,leading to a higher imaxata higher Emax,amore positive of Epassive,iand a narrower passive region.Appearance of one ormore anodic current peaks in bicarbonate solutionsw ith high concentration of sulfate comes from the reactivation of several dissolution or oxidation reactions.According to Fig.20, reactions(1)to(11)may happen atdifferentelectrode potentials. The results show thatcorrosion rate of iron in bicarbonate solutions can be significantly high if ahigh concentration of sulfate is present.
5 Conc lusions
In the absence of sulfate or in the presence of a low concentration of sulfate in dilute bicarbonate solutions,iron is in passive state with corrosion potential of(-0.225±0.005)V.High electrochemical impedanceand low corrosion ratewere obtained.No obviousactive-passive transitionwasshown on anodic polarization curves.
In the presence of a high concentration of sulfate in dilute bicarbonate solutions,iron is in active dissolution state w ith corrosion potential of(-0.790±0.010)V.Low values of electrochem ical impedance,high corrosion rate,and typical activepassive transition on anodic polarization curveswere observed and related to the sulfate concentrations.Transition of open circuit state from passivity to active dissolution occursata lower sulfate concentration in adeaerated solution than in an aerated solutions.
Anodic polarization curves of iron in dilute bicarbonate solutions changew ith sulfate concentration.In the presence of a high concentration of sulfate,anodic polarization curvesexhibit current peaksas the resultsof theactivation of iron by sulfate ions.
(1)Fraunhofer,V.J.A.Corrosion Sci.1970,10,245.doi:10.1016/ S0010-938X(70)80084-1
(2)Thomas,J.G.N.;Nurse,T.J.;Walker,R.Br.Corros.J.1970,5, 87.doi:10.1179/000705970798324829
(3)Pourbaix,M.Lectureson ElectrochemicalCorrosion;Plenum Press:New York-London,1973.
(4)Davis,D.H.;Burstein,G.T.Corrosion 1980,36,416.doi: 10.5006/0010-9312-36.8.416
(5)Jelink,J.;Neufeld,P.Corrosion Sci.1980,20,489.doi:10.1016/ 0010-938X(80)90066-9
(6)Sutcliffe,J.M.;Fessler,R.R.;Boyd,W.K.;Parkins,R.N. Corrosion 1972,28,313.doi:10.5006/0010-9312-28.8.313
(7)Stiksma,J.;Bradford,S.A.Corrosion 1985,41,446.doi: 10.5006/1.3583825
(8)Castro,E.B.;Valentini,C.R.;Moina,C.A.;Vilche,J.R.; Arvia,A.J.Corrosion Sci.1986,26,781.doi:10.1016/0010-938X(86)90063-6
(9)Rangel,C.M.;Leitao,R.A.Electrochim.Acta 1989,34,255. doi:10.1016/0013-4686(89)87094-X
(10)Videm,K.;Koren,A.M.Corrosion 1993,49,746.doi:10.5006/ 1.3316127
(11)Simard,S.;Drogow ska,M.;Menard,H.;Brossard,L.J.App. Electrochem.1997,27,317.doi:10.1023/A:1018484814627
(12)Simard,S.;Menard,H.J.App.Electrochem.1998,28,151.
(13)Legrand,L.;Savoye,S.;Chausse,A.;Messina,R.Electrochim. Acta 2000,46,111.doi:10.1016/S0013-4686(00)00563-6
(14)A lves,V.A.;Brett,C.M.A.Electrochim.Acta 2002,47, 2081.doi:10.1016/S0013-4686(02)00077-4
(15)Lu,Z.P.;Huang,C.B.;Huang,D.L.;Yang,W.Corrosion Sci. 2006,48,3049.doi:10.1016/j.corsci.2005.11.014
(16)Acosta,C.A.;Salvarezza,R.C.;Videla,H.A.;Arvia,A.J. Corrosion Sci.1985,25,291.doi:10.1016/0010-938X(85) 90108-8
(17)Zhao,J.M.;Zuo,Y.;Xiong,J.P.;Tian,L.P.Corros.Sci.Prot. Technol.2001,13,77.[赵景茂,左 禹,熊金平,田连朋.腐蚀科学与防护技术,2001,13,77.]
(18)Vatankhah,G.;Drogowska,M.;Menard,H.J.App. Electrochem.1998,28,173.
(19)Haruna,T.;Domoto,K.;Shibata,T.Corrosion Eng.2002,51, 350.doi:10.3323/jcorr1991.51.350
(20)El-Naggar,M.M.App.Surf.Sci.2006,52,6179.
(21)Lu,Z.P.;Chen,J.M.Br.Corros.J.2000,35,224.doi:10.1179/ 000705900101501281
(22)Waard,C.;M illiams,D.E.Corrosion 1975,31,177.doi: 10.5006/0010-9312-31.5.177
(23)Turgoose,S.;Cottis,R.A.;Lawson,K.Modeling of Electrode Processesand Surface Chemistry in Carbon Dioxide Containing Solutions.In ComputerModeling in Corrosion,ASTM STP 1154;Munn,R.S.Ed.;American Society for Testing and Materials:Philadephia,1992;pp 67-81.
(24)Linter,B.R.;Burstein G.T.Corrosion Sci.1999,41,117.doi: 10.1016/S0010-938X(98)00104-8
(25)Han,J.B.;Zhang,J.S.;Careya,J.W.Int.J.Greenhouse Gas Control2011,5,1680.doi:10.1016/j.ijggc.2011.08.003
(26)Marsh,G.P.Nucl.Energy 1982,21,253.
(27)Xiao,F.;Wang,J.;Guo,Y.H.;Wang,Z.M.;Su,R.Uranium Geol.2011,27,185.[肖 丰,王 驹,郭永海,王志明,苏锐.铀矿地质,2011,27,185.]
(28)Yang,J.F.;Dong,J.H.;Ke,W.Acta Metall.Sin.(Chin.Ed.) 2011,47,1321.[阳靖峰,董俊华,柯 伟.金属学报,2011, 47,1321.]
(29)Andrade,C.;Alonso,C.;Sarria,J.Cem.Concr.Compos.2002, 24,55.doi:10.1016/S0958-9465(01)00026-9
(30)Deodeshmukh,V.;Venugopal,A.;Chandra,D.;Yilmaz,A.; Daemen,J.;Jones,D.A.;Lea,S.;Engelhard,M.Corrosion Sci. 2004,46,2629.doi:10.1016/j.corsci.2004.03.007
(31)Mao,X.;Liu,X.;Revie,R.W.Corrosion 1994,50,651.doi: 10.5006/1.3293540
(32)Liu,X.;Mao,X.Scr.Metall.Mater.1995,33,145.doi:10.1016/ 0956-716X(95)00112-9
(33)Guo,H.;Du,C.W.;Li,X.G.;Chen,Y.X.Equip.Environ.Eng. 2007,4,40.[郭 昊,杜翠薇,李晓刚,陈云祥.装备环境工程,2007,4,40.]
(34)Zhou,J.L.;Li,X.G.;Du,C.W.;Li,Y.L.;Li,T.;Pan,Y.Acta Metall.Sin.(Chin.Ed.)2010,46,251.[周建龙,李晓刚,杜翠薇,李云玲,李 涛,潘 莹.金属学报,2010,46,251.]
(35)Eliyan,F.F.;Mahdi,E.;A lfantazi,A.Corrosion Sci.2012,58, 181.doi:10.1016/j.corsci.2012.01.015
(36)Hu,J.Y.;Cao,S.A.;Xie,J.L.Acta Phys.-Chim.Sin.2012,28, 1153.[胡家元,曹顺安,谢建丽.物理化学学报,2012,28, 1153.]doi:10.3866/PKU.WHXB201203022
(37)Parkins,R.N.;Zhou,S.Corrosion Sci.1997,39,175.doi: 10.1016/S0010-938X(97)89248-7
(38)Heuer,J.K.;Stubins,J.F.Corrosion Sci.1999,41,1231.doi: 10.1016/S0010-938X(98)00180-2
(39)Bockris,J.O.M.;Drazic,D.Electrochim.Acta 1962,7,293. doi:10.1016/0013-4686(62)87007-8
(40)Shipley,H.J.;Gao,Y.;Kan,A.T.;Tomson,M.B.The Mobilization of Metalsand Inorganic Compoundsduring Resuspension of Anoxic Sediment.In the11th Annual InternationalPetroleum EnvironmentalConference, Albuquerque,NM,USA,October12-15,2004.
(41)Castro,E.B.;Vilche,J.R.J.App.Electrochem.1991,21,543. doi:10.1007/BF01018608
(42)Zeng,Y.M.;Luo,J.L.;Norton,P.R.Electrochim.Acta 2004, 49,703.doi:10.1016/j.electacta.2003.09.024
(43)Florianovich,G.M.;Sokolova,L.A.;Kolotyrkin,Y.M. Electrochim.Acta 1967,12,879.doi:10.1016/0013-4686(67) 80124-5
(44)Pound,B.G.;W right,G.A.;Sharp,R.M.Corrosion 1989,45, 386.doi:10.5006/1.3582033
(45)Szklarska-Smialowska,S.;M rowczynski,G.Br.Corros.J. 1975,10,187.doi:10.1179/000705975798320486
(46)M ills,P.;Sullivan,J.L.JournalofPhysicsD:Applied Physics 1983,16,723.doi:10.1088/0022-3727/16/5/005
(47)Moulder,J.F.;Stickle,W.F.;Sobol,P.E.;Bomben K.D. Handbook ofX-ray Photoelectron Spectroscopy;Perkin-Elmer Corporation:Eden Prairie,M innesota,1995.
(48)Tan,B.J.;K labunde,K.J.;Sherwood,P.M.A.Chem.Mater. 1990,2,186.doi:10.1021/cm00008a021
(49)Yamashita,T.;Hayes,P.App.Surf.Sci.2008,254,2441.doi: 10.1016/j.apsusc.2007.09.063
(50)Allen,G.C.;Curtis,M.T.;Hooper,A.J.;Tucker,P.M.J.Chem. Society Dalton Transactions1974,14,1525.
(51)Pitzer,K.S.;Peiper J.C.J.Phys.Chem.1980,84,2396.doi: 10.1021/j100456a011
(52)Corrosion Mechanisms in Theory and Practice,3rd ed.;Marcus, P.Ed.;CRCPress:Boca Raton,2011.
(53)Chen,J.J.;Lu,Z.P.;Xiao,Q.;Xia,X.F.;Xia,S.;Yao,M.Y.; Zhou,B.X.ECSTrans.2014,59,335.doi:10.1149/ 05901.0335ecst
Effec ts o f Su lfate Ions on Anod ic Disso lu tion and Passivity o f Iron in Sligh tly A lkaline So lu tions
CHEN Jun-Jie XIAO Qian LÜZhan-Peng*AHSAN Ejaz XIAXiao-Feng LIU Ting-Guang
(1Institute ofMaterials Science,SchoolofMaterials Science and Engineering,ShanghaiUniversity,Shanghai200072,P.R.China;2State Key LaboratoryofAdvanced SpecialSteels,ShanghaiUniversity,Shanghai200072,P.R.China)
The effects ofsulfate concentration on the open circuitstate and anodic polarization behavior of iron in dilute bicarbonate solutions were investigated using immersion tests,electrochem icalmeasurements, and surface analysis techniques.In the absence ofsulfate or in the presence ofa low concentration ofsulfate in dilute bicarbonate solutions,iron was in a passive state,with a corrosion potentialof(-0.225±0.005)V.Ahigh electrochem icalim pedance and low corrosion rate were obtained.No obvious active-passive transition was observed in the anodic polarization curves.In the presence of a high concentration of sulfate in dilute bicarbonate solutions,iron was in an active dissolution state,w ith a corrosion potentialof(-0.790±0.010)V. A low electrochem icalimpedance,high corrosion rate,and typicalactive-passive transition in anodic polarization curveswere observed and related to the sulfate concentration.In the presence ofa high concentration ofsulfate, the anodic polarization curves showed currentpeaks as a resultof iron activation by sulfate ions.Sulfate ions ofsufficiently high concentration in solutions degraded previously formed oxide layers on iron or transformed oxide layers in bicarbonate solutions.The transition of the open circuitstate from passivity to active dissolution occurs ata lower sulfate concentration in a deaerated solution than in an aerated solution.
O646;TG171
icle]
10.3866/PKU.WHXB201504032 www.whxb.pku.edu.cn
Received:February 2,2015;Revised:April3,2015;Published onWeb:April3,2015.
∗Corresponding author.Email:zplu@shu.edu.cn;Tel:+86-21-56336107.
The projectwassupported by the InternationalCooperative ProjectSponsored by Scienceand Technology Commission of ShanghaiMunicipality,
China(13520721200),Specialized Research Fund for the DoctoralProgram of Higher Education,China(20123108110021),and ShanghaiPujiang Project,China(12PJ1403600).
上海市科委国际科技合作项目(13520721200),高等学校博士学科点专项科研基金博导类项目(20123108110021)及上海市浦江人才计划项目(12PJ1403600)资助
©EditorialofficeofActa Physico-Chim ica Sinica