铂钌团簇活化甲醇C―H和O―H键的理论研究
2015-12-29赵俊凤孙小丽李吉来黄旭日
赵俊凤 孙小丽 李吉来 黄旭日
(吉林大学理论化学研究所,长春130023)
铂钌团簇活化甲醇C―H和O―H键的理论研究
赵俊凤 孙小丽 李吉来*黄旭日*
(吉林大学理论化学研究所,长春130023)
采用密度泛函理论研究了PtnRum(n+m=3,n≠0)团簇活化甲醇C―H和O―H键的反应活性和机理.分别给出以O―H和C―H键活化为初始步骤的势能曲线.计算结果表明反应是以C―H键的活化为初始步骤;三种团簇与甲醇反应的活性顺序为Pt2Ru>Pt3>PtRu2.前线轨道分析表明PtnRum团簇活化初始的C―H和O―H键的活化过程是质子转移(PT).此外还讨论了溶剂化对反应的影响.本研究可为C―H键和O―H键的活化提供更深入的理解,为甲醇活化反应催化剂选择以及其使用条件的优化提供新思路.
密度泛函理论;团簇;甲醇;活性;质子转移
1 引言
C―H键活化和功能化研究在有机合成反应、分子转化过程以及构建复杂生物结构方面具有重要作用.1-19O―H键的活化在化学和化工生产中也十分重要,在实验和理论上也有一些研究.20,21因此对C―H键和O―H键活化进行深入的理论研究非常有必要,尤其是对于同时含有C―H键和O―H键的物质,因为活化的初始步骤存在选择性问题,醇类物质作为研究对象是一个很好的选择.甲醇是脂肪醇中最小的分子,它不仅是重要的工业合成原料,22而且作为液态燃料,易贮存和运输,原料丰富、价格低廉,亦具有较高的反应活性,可以作为氢的替代燃料.甲醇的一个重要应用就是在直接甲醇燃料电池(DMFC)上,DMFC具有能量密度高、运行温度低、环境友好、使用方便等优点,它作为未来清洁的动力能源受到人们广泛的关注,其广阔的应用前景引起了国内外的高度重视.23-27
目前DMFC阳极催化剂主要采用贵金属催化剂,如Pt、Pd、Au等,28-30当然也有一些包括金属碳化物和过渡金属氧化物在内的非贵金属催化剂,例如碳化钨、某些钙钛矿类氧化物、NiZr合金等.31-33虽然非贵金属催化剂的使用能够大大降低电池成本,但其催化效率却并不理想.Pt基催化剂仍然是迄今为止甲醇氧化最有效的催化剂.34因为其具有未填满的d电子轨道,Pt表面极易吸附反应物,强度适中,并且有利于形成高活性的中间产物;另外,它还具有耐高温、抗氧化、耐腐蚀等优良特性.因此基于Pt基催化剂的改性研究以提高其催化效率更加具有现实意义.目前对Pt基催化剂的改性研究,主要在两个方面:(1)减少Pt的载量,采用一些高比表面积的载体,如分子筛或碳纳米管,提高其利用率;35,36(2)向Pt中加入一种或几种金属得到二元或多元催化剂,从而改进催化剂的性能.有较高反应活性的二元Pt基催化剂有:PtRu、37-39PtAu、40,41PtMo42和PtSn43,44等.其中PtRu二元催化剂是迄今为止最有效的DMFC阳极催化剂.45这些高效催化剂催化的反应引起了大量研究者的注意,它们微观结构特点和独特的物理化学性质也被广泛研究.46,47在PtRu二元催化剂中Ru的作用主要有两方面:一是Ru将部分d电子传递给Pt,减弱Pt和CO之间相互作用,同时使吸附的含碳中间物中的C原子上正电荷增加,使其更容易受到水分子的亲核攻击;二是增加催化剂表面含氧物种覆盖度.PtRu二元催化剂中的Pt主要是还原态的,而Ru主要是氧化态的.少量的还原态Ru存在于催化剂的体相而不是表面.以往的研究也证明PtRu二元催化剂中Pt原子是主要的反应活性中心,48,49单纯的Ru对甲醇没有催化氧化活性,Ru的加入对Pt催化剂性能有较好的改善.50目前关于铂团簇的实验合成效率低,分离困难,关于团簇、壳层结构的实验信息也还很有限,因此,理论研究成为当前研究的热点之一.系统考察反应的内在反应机制对提高Pt基催化剂的反应活性以及改善DMFC的性能无疑具有重要意义.
基于以上动机,我们对PtnRum(n+m=3,n≠0)小团簇模型与甲醇的气相反应进行系统的研究,本文中只讨论以Pt原子为活性中心的反应.鉴于甲醇脱氢机制以往也有报道,51本文研究重心不再单纯重复探讨甲醇活化的具体步骤和过程,研究的重点集中在:(1)甲醇活化过程中PtnRum团簇活化C―H键和O―H键的区别以及电子转移过程;(2)溶剂对甲醇活化反应的影响,分别考虑了气相、水、乙腈和介电常数ε=4时四种情况对反应的影响.本文对C―H键和O―H键的活化机制进行了详细的理论探索,辅以电子结构分析,以期揭示其反应本质,为甲醇活化反应的催化剂设计提供理论指导.
2 计算方法
本文所有计算采用Gaussian 09程序包.52采用B3LYP方法,53对Ru和Pt选用了双ζ价电子基组和相应的Los Alamos有效核势54,55LANL2DZ基组,对C、H和O原子选用6-31G(d)基组确定势能面上的各驻点,并在相同水平下进行了频率计算,得到热力学信息.本文所有过渡态都有唯一虚频,并通过振动模式分析进一步确认了过渡态的真实性.采用内禀坐标(IRC)56计算来验证反应路径的正确连接.为了得到更准确的能量信息,采用B3LYP/def2-QZVPP理论水平计算单点能构建精确的势能面.为了掌握电子转移细节,加深对反应的理解,对在最小能量路径上选择代表性点进行准束缚分子轨道(QRO)57-64分析,分子轨道图使用Chimera软件65绘制.
尽管近期的研究表明没有一个单一的密度泛函理论方法可以适用于多个体系,但是方法的基准研究表明对于过渡金属体系中涉及金属氢键(M―H)、金属碳键(M―C)、金属氧键(M―O),B3LYP泛函具有很强的适用性.66-70因此,基于本文的研究重点意在给出一个定性的趋势,这个方法的结果是可以接受的.

表1 PtnRum(n+m=3,n≠0)不同自旋态下的相对能量Table 1 Relative energiesof different spin statesof PtnRum(n+m=3,n≠0)
3 结果与讨论
表1中给出Pt3、Pt2Ru和PtRu2团簇模型在不同自旋态下的相对能量,从表中可以看出:(1)Pt3团簇低自旋态稳定,单重态(S=0)能量最低,三重态(S=1)次之,能量为67.0 kJ·mol-1;(2)在纯Pt团簇中掺杂Ru形成Pt2Ru团簇后,五重态(S=2)能量最低,三重态(S=1)能量次之,三重态较五重态能量高32.7 kJ·mol-1;(3)PtRu2团簇高自旋态稳定,五重态(S=2)和七重态(S=3)能量相对最低.对比三种团簇Pt3、Pt2Ru和PtRu2基态时的自旋状态,可以看出随着Ru原子取代数目的增加,团簇的稳定状态趋于高自旋.本文中选取Pt3、Pt2Ru和PtRu2各自能量较低的两个自旋态进行研究.图1是在B3LYP水平下优化得到PtnRum(n+m=3,n≠0)几何构型.
PtnRum(n+m=3,n≠0)团簇活化甲醇中的O―H键和C―H键,M 1机制:以O―H键活化为起始步骤,再进行C―H键的活化.如示意图1所示,O―H键活化有两种方式:(1)Path A中单个金属原子直接参与O―H键活化,TSOa为相应过渡态;(2)Path B中两个金属原子同时参与O―H键活化,TSOb为相应过渡态.C―H键活化时仅存在单个金属原子参与活化的反应机制,TSOaHCa为相应过渡态.M 2机制:以C―H键活化为起始步骤,再进行O―H键的活化.如示意图2所示,C―H键活化方式为单个金属原子参与活化反应,TSCa为相应过渡态.O―H键活化有两种方式:(1)Path A中单个金属原子直接参与O―H键活化;(2)Path B中两个金属原子同时参与O―H键活化,TSCaOb为相应过渡态.其中O―H键活化的Path A过程中Pt3团簇与Pt2Ru、PtRu2团簇存在不同之处,示意图2中TSCaHOa过渡态对应的是Pt3团簇活化O―H键的Path A过程;TSCaOa过渡态对应的是Pt2Ru、PtRu2团簇活化O―H键的Path A过程.在本文中,我们把分离反应物(SR)即PtnRum+CH3OH作为初始态,金属中心与甲醇的O作用形成反应复合物RC_O,对应M 1机制;金属中心与甲醇的甲基H相互作用形成反应复合物RC_C,对应M 2机制.
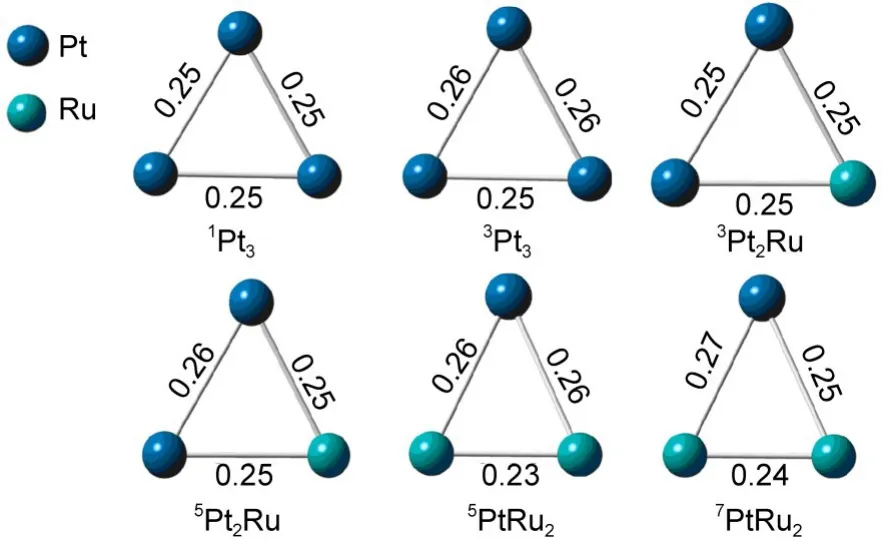
图1 在B3LYP水平下优化得到PtnRum(n+m=3,n≠0)几何构型Fig.1 Optim ized geometriesof PtnRum(n+m=3,n≠0)at B3LYP levelof theory

示意图1 PtnRum与CH3OH以O―H键活化为初始步骤(M 1)的反应Scheme1 Reaction of PtnRumw ith CH3OH starting from O―H bond activation(M 1)
3.1 M1机制
图2A为Pt3与甲醇反应M 1机制的焓变势能面.甲醇分子通过氧原子与Pt原子结合,形成络合物RC_O,1,3RC_O的能量分别为-70.3和-62.0 kJ·mol-1. O―H键断裂通过过渡态TSOa与TSOb实现且分别生成中间体I1和I2.TSOa与TSOb单重态能量相差较大,1TSOa的能量仅为20.5 kJ·mol-1,1TSOb的能量为55.3 kJ·mol-1;三重态时两个过渡态能量接近,3TSOa与3TSOb分别为25.1和27.6 kJ·mol-1,单个Pt原子参与活化的活化机制依然占优势.Pt3活化O―H键时,采用单个Pt原子参与活化的活化机制(经过1TSOa)较为有利.I1可以通过过渡态TSOaHd将H原子转移到相邻Pt原子上形成I2,从1,3TSOaHd的能量(-49.4vs-45.2 kJ·mol-1)来看,H原子在两个Pt原子上转移的过程在动力学和热力学上都是非常容易的.从I2到I3为C―H键活化过程,1TSOaHCa的能量(-70.8 kJ·mol-1)明显低于3TSOaHCa的能量(-25.1 kJ·mol-1).综合整个势能面,显然最初O―H键的断裂是反应的决速步骤.综合整个势能面,反应最优通道为1RC_O➝1TSOa➝1I1➝1TSOaHd➝1I2➝1TSOaHCa➝1I3.图2B、2C给出Pt2Ru、PtRu2与甲醇反应M 1机制的焓变势能面,与Pt3相似,活化O―H键时,采用单个Pt原子参与活化的活化机制有利,O―H键的断裂是反应的决速步骤.

示意图2 PtnRum与CH3OH以C―H键活化为初始步骤(M 2)的反应Scheme2 Reaction of PtnRumw ith CH3OH starting from C―H bond activation(M 2)
在M 1机制中,活化O―H键,Pt2Ru和PtRu2比Pt3活性高,活化C―H键时则相反.综合Pt3、Pt2Ru和PtRu2与甲醇反应的最优通道的势能面(图2),Pt2Ru团簇与甲醇的反应的活性最高.图S1、S2和S3(见Supporting Information)为M 1机制中涉及的几何结构.
3.2 M 2机制
图3A是Pt3与甲醇反应M 2机制的焓变势能面. RC_C是甲醇分子与Pt3团簇形成的络合物,甲醇中甲基上的H原子与Pt原子相互作用.1,3RC_C的相对能量分别为-27.2和-32.7 kJ·mol-1.尽管RC_C与RC_O相比是亚稳定的构型,但是这种亚稳定的结构在Pt(111)面甲醇吸附的过程中是存在的.71C―H键活化采用单个Pt原子活化的反应机制,1,3TSCa的能量分别为-18.8和-17.6 kJ·mol-1.活化C―H键中单重态和三重态在能量上互相竞争,生成中间体I4.从中间体I4开始O―H的活化有两种反应机制: (1)I4到I5,将H原子转移到相邻Pt原子上,H转移后再活化O―H键,O―H键的活化是单个Pt原子参与活化反应,经过过渡态TSCaHOa,1,3TSCaHOa的能量分别为-14.7和28.1 kJ·mol-1,伴随着O―H键的断裂,Pt―H键生成,得到中间体I6;(2)I4中O―H键的活化是两个Pt原子同时参与活化反应的,经过过渡态TSCaOb,1,3TSCaOb的能量分别为-31.4和24.3 kJ·mol-1,生成中间体I7,从图S4(见Supporting Information)中1,3TSCaOb结构中可以看出,过渡态中有一个Pt…C…O…H…Pt的五元环,不同的是1TSCaOb中伴随着O-H键的活化,第二个C―H键同时被活化.O―H键的活化b过程比a过程有利.综合整个势能面,M 2机制中单重态反应过程中反应复合物、中间体和过渡态的能量都低于SR,最优反应途径为1RC_C➝1TSCa➝1I4➝1TSCaOb➝1I7.
对比图2A和图3A所示的两个反应过程势能面图可以看出,Pt3团簇与甲醇反应以C―H键活化为初始步骤的反应路径比以O―H键活化为初始步骤的反应路径有利.这与Greeley和Mavrikakis72的研究结果是一致的.值得一提的是,M 2机制中O―H键的活化过程两个Pt原子同时参与活化的过程比单个Pt原子参与活化的过程有利,而M 1机制中情况却正好相反,可以推测O―H键活化方式与其被活化的顺序有关.

图2 B3LYP水平下气相Pt3(A),Pt2Ru(B)和PtRu2(C)与CH3OH反应的以O―H键活化为初始步骤(M 1)的ΔH(g)势能面Fig.2 Potentialenergy surface ofΔH(g)for the reactionsof Pt3(A),Pt2Ru(B),and PtRu2(C)w ith CH3OH starting from O―H bond activation(M 1)at the B3LYP levelof theory in gasphase
图3B、3C是Pt2Ru和PtRu2与甲醇反应M 2机制的焓变势能面.反应复合物RC_C,C―H键活化通过过渡态TSCa生成中间体I4.I4中O―H键活化有两种方式:(a)单个Pt原子参与O―H键活化,通过过渡态TSCaOa生成中间体I6;(b)两个金属原子同时参与O―H键活化,通过过渡态TSCaOb生成中间体I5.从势能面可以看出后者较前者有利,即M 2机制中O―H键活化采用两个金属原子同时参与活化的反应机制有利,这与Pt3团簇相同,进一步说明O―H键活化方式与其被活化的顺序有关.
在M 2机制中,Pt2Ru和Pt3活化甲醇的C―H和O―H键,反应复合物、中间体和过渡态的能量都低于对应的SR,Pt3和Pt2Ru团簇与甲醇的反应活性高于PtRu2.图S4、S5和S6(见Supporting Information)为M 2机制涉及的几何结构.
综上,Pt3、Pt2Ru和PtRu2活化甲醇都是M 2机制占优势,即反应以C―H键活化为初始步骤.O―H键的活化是反应的决速步骤.O―H键活化方式与O―H活化的先后顺序有关:M 1机制中是单个Pt原子参与活化有利;M 2机制中C―H键先被活化,再活化O―H键,则是两个金属原子同时参与活化有利.综合M 1和M 2,三种团簇与甲醇活性最高的为Pt2Ru.

图3 B3LYP水平下气相Pt3(A),Pt2Ru(B)和PtRu2(C)与CH3OH反应以C―H键活化为反应的初始步骤(M 2)的ΔH(g)势能面Fig.3 Potentialenergy surface ofΔH(g)for the reaction of Pt3(A),Pt2Ru(B),and PtRu2(C)w ith CH3OH starting from C―H bond activation(M 2)at the B3LYP levelof theory in gasphase
3.3 溶剂化作用
表S1、S2和S3(见Supporting Information)分别列出了Pt3、Pt2Ru、PtRu2在介电常数(ε=4)、乙腈溶剂(ε=36.6)和水(ε=78.4)条件下的相对焓变,从表中可以看出Pt3体系在溶剂下能量比气相下的高,并且从反应物到产物这样的变化会更加明显,过渡态的能垒在溶剂下略降低.在Pt2Ru体系中总体上溶剂中能量比气相的高,个别的结构能量有稍微的降低,随着Ru的加入,在PtRu2体系中,溶剂对反应的影响趋于有利的方向发展,我们可以推测,随着Ru的比例的增加,极性溶剂对催化甲醇的反应变得有利,尤其是在ε=36.6.所以Ru的加入可以在某种程度上降低对环境的选择性.
3.4 电子结构分析
图4为Pt3(S=0,1)、Pt2Ru(S=1,2)和PtRu2(S=2, 3)的前线分子轨道图.在Pt3中,单重态Pt原子的5d轨道均为双占据轨道,三个Pt之间都是σ键;三重态时,两个单电子分别占据π*Pt―Pt和σ*Pt―Pt―Pt,很明显三重态其中两个Pt之间多了一个π键.在Pt2Ru中,三重态时,两个Pt的5个d轨道为双占据轨道,两个单电子占据在Ru的dxz和dz2的轨道.五重态时,单电子占据在Ru的dxz、dyz、dxy和σ*Pt―Pt―Ru反键轨道,Pt原子d轨道全部为双占据轨道.在PtRu2中,五重态时5PtRu2四个单电子占据Ru的dxz、dxy和dz2的轨道和π*Ru―Ru反键轨道,Pt原子的5个d轨道为双占据轨道.七重态时单电子占据Ru的三个d轨道和反键轨道σ*Ru―Ru、π*Ru―Ru,Pt原子d轨道全部为双占据轨道.从这些可以看出,从Pt3→Pt2Ru→PtRu2,Ru替换Pt的时候只替换有单占据轨道的Pt,即掺杂Ru的Pt催化剂单电子都集中在Ru上,这也可能是随着Ru的增加PtnRum(n+m=3,n≠0)基态趋向于高自旋的原因.形式上并未改变Pt的价态.
图S7(见Supporting Information)是Pt3(S=0,1)首先活化O―H键的前线轨道图,从1RC_O、1TSOa和1I1的前线轨道图中可以看出,参与O―H键活化的Pt原子五个d轨道始终为双占据轨道,从1RC_O到1I1,H转移到Pt上,但电子没有发生转移;从3RC_O、3TSOa和3I1的前线轨道图中可以看出,两个单电子始终占据σ*Pt―Pt―Pt和π*Pt―Pt反键轨道,参与O―H键活化的Pt原子五个d轨道始终为双占据轨道.说明初始O―H键活化过程是质子转移(PT)的过程.图S8是Pt3(S=0,1)首先活化C―H键的前线轨道图,与活化O―H键的前线轨道图相似,从RC_C、TSCa和I4的前线轨道图中可以看出,参与C―H键活化的Pt原子五个d轨道始终为双占据轨道,H转移到Pt上,电子没有发生转移.说明初始C―H键活化过程也是质子转移的过程.图S9-S12(见Supporting Information)是Pt2Ru(S=1,2)和PtRu2(S=2,3)首先活化O―H键和C―H键的前线轨道图,与Pt3(S=0,1)活化O―H键和C―H键的过程相似,参与活化的Pt原子五个d轨道始终为双占据轨道,H转移到Pt上,电子没有发生转移,说明初始O―H键和C―H键活化过程是质子转移的过程.

图4 Pt3(S=0,1),Pt2Ru(S=1,2)和PtRu2(S=2,3)的前线分子轨道(FMO)图Fig.4 Schematic frontiermolecular orbital(FMO) diagram of Pt3(S=0,1),Pt2Ru(S=1,2),and PtRu2(S=2,3)
4 结论
本文采用密度泛函理论,在B3LYP水平下研究了PtnRum(n+m=3,n≠0)团簇与甲醇的活化反应.对比分别以O―H键和C―H键活化为初始步骤的反应活性并构建了反应势能面,分析了反应过程的电子结构,讨论了溶剂化(水溶液、乙腈和介电常数ε= 4)对甲醇活化反应的影响.通过详尽的理论计算得到以下结论.
(1)PtnRum团簇活化甲醇应以C―H的活化为初始步骤.
(2)O―H键的活化是反应的决速步骤.
(3)O―H键活化方式与活化的先后顺序有关.如果O―H键首先被活化,一个Pt原子参与活化有利;如果先活化C―H键,再活化O―H键,则两个金属原子同时参与活化有利.
(4)前线轨道分析表明第一步活化C―H和O―H键都是质子转移.
(5)三种团簇与甲醇反应的活性顺序为Pt2Ru>Pt3>PtRu2,说明在纯Pt团簇中适当掺杂Ru能提高其反应活性.
(6)Pt3团簇在气相下反应活性比溶剂下反应活性大;掺杂Ru后,增加溶剂极性对反应的影响趋于有利的方向发展;随着Ru比例的增加,这种有利的趋势越来越明显.
本文的研究为C―H键和O―H键的活化提供更深入的理解,对PtRu二元金属团簇与甲醇活化反应的反应机理、活化性能的改良具有重要意义,可为直接甲醇燃料电池中高效催化剂的设计提供理论参考.
Suppo rting In fo rm ation:Enthalpies,optimized geometries,and schematic FMO diagram involved in the reaction of PtnRum(n+m=3,n≠0)w ith CH3OH have been included.This information is available free of charge via the internet at http:// www.whxb.pku.edu.cn.
(1)de Visser,S.P.;Shaik,S.J.Am.Chem.Soc.2003,125, 7413.doi:10.1021/ja034142f
(2)Schwarz,H.;Schröder,D.Pure Appl.Chem.2000,72,2319.
(3)Schwarz,H.Angew.Chem.Int.Edit.2011,50,10096.doi: 10.1002/anie.201006424
(4)Sun,X.L.;Li,J.L.;Huang,X.R.;Sun,C.C.Curr.Inorg. Chem.2012,2,64.doi:10.2174/1877944111202010064
(5)Li,J.L.;Zhang,X.;Huang,X.R.Phys.Chem.Chem.Phys. 2012,14,246.doi:10.1039/C1CP22187F
(6)Li,J.L.;Geng,C.Y.;Huang,X.R.;Zhang,X.;Sun,C.C. Organometallics2007,26,2203.doi:10.1021/om070039d
(7)Li,J.L.;Wu,X.N.;Schlangen,M.;Zhou,S.D.;González-Navarrete,P.;Tang,S.Y.;Schwarz,H.Angew.Chem.Int.Edit. 2015,doi:10.1002/anie.201412441.
(8)Shaik,S.;de Visser,S.P.;Ogliaro,F.;Schwarz,H.;Schröder,D. Curr.Opin.Chem.Biol.2002,6,556.doi:10.1016/S1367-5931 (02)00363-0
(9)Ye,S.;Neese,F.Curr.Opin.Chem.Biol.2009,13,89.doi: 10.1016/j.cbpa.2009.02.007
(10)Ye,S.;Neese,F.Proc.Natl.Acad.Sci.U.S.A.2011,108, 1228.doi:10.1073/pnas.1008411108
(11)Neese,F.J.Inorg.Biochem.2006,100,716.doi:10.1016/j. jinorgbio.2006.01.020
(12)Geng,C.Y.;Ye,S.;Neese,F.Angew.Chem.Int.Edit.2010,49, 5717.doi:10.1002/anie.v49:33
(13)Geng,C.Y.;Li,J.L.;Huang,X.R.;Liu,H.L.;Li,Z.;Sun,C. C.J.Comput.Chem.2008,29,686.
(14)Decker,A.;Rohde,J.U.;K linker,E.J.;Wong,S.D.;Que,L.; Solomon,E.I.J.Am.Chem.Soc.2007,129,15983.doi: 10.1021/ja074900s
(15)Shaik,S.;Kumar,D.;de Visser,S.P.;Altun,A.;Thiel,W. Chem.Rev.2005,105,2279.doi:10.1021/cr030722j
(16)Shaik,S.;Cohen,S.;Wang,Y.;Chen,H.;Kumar,D.;Thiel,W. Chem.Rev.2009,110,949.
(17)Schöneboom,J.C.;Cohen,S.;Lin,H.;Shaik,S.;Thiel,W. J.Am.Chem.Soc.2004,126,4017.doi:10.1021/ja039847w
(18)Kwon,Y.H.;Kim,S.C.;Lee,S.Y.Macromolecules2009,42, 5244.doi:10.1021/ma900781c
(19)Martínez-Huerta,M.V.;Rodríguez,J.L.;Tsiouvaras,N.;Peña, M.A.;Fierro,J.L.G.;Pastor,E.Chem.Mater.2008,20, 4249.doi:10.1021/cm703047p
(20)M ichel,C.;Goltl,F.;Sautet,P.Phys.Chem.Chem.Phys.2012, 14,15286.doi:10.1039/c2cp43014b
(21)Ranea,V.A.;M ichaelides,A.;Ramírez,R.;deAndres,P.L.; Vergés,J.A.;King,D.A.Phys.Rev.Lett.2004,92,136104. doi:10.1103/PhysRevLett.92.136104
(22)Usami,Y.;Kagawa,K.;Kawazoe,M.;Yasuyuki,M.;Sakurai, H.;Haruta,M.Appl.Catal.A-Gen.1998,171,123.doi: 10.1016/S0926-860X(98)00082-9
(23)Hamnett,A.Catal.Today 1997,38,445.doi:10.1016/S0920-5861(97)00054-0
(24)Childers,C.L.;Huang,H.L.;Korzeniewski,C.Langmuir1999, 15,786.doi:10.1021/la980798o
(25)Xu,C.;Wang,R.;Chen,M.;Zhang,Y.;Ding,Y.Phys.Chem. Chem.Phys.2010,12,239.doi:10.1039/B917788D
(26)Hernández-Fernández,P.;Montiel,M.;Ocón,P.;Fierro,J.L. G.;Wang,H.;Abruña,H.D.;Rojas,S.J.Power Sources2010, 195,7959.doi:10.1016/j.jpowsour.2010.06.009
(27)Wen,Z.;Liu,J.;Li,J.Adv.Mater.2008,20,743.
(28)Li,Y.;Tang,L.;Li,J.Electrochem.Commun.2009,11,846. doi:10.1016/j.elecom.2009.02.009
(29)Zhao,Y.;Zhan,L.;Tian,J.;Nie,S.;Ning,Z.Electrochim.Acta 2011,56,1967.doi:10.1016/j.electacta.2010.12.005
(30)Santhosh,P.;Gopalan,A.;Lee,K.P.J.Catal.2006,238, 177.doi:10.1016/j.jcat.2005.12.014
(31)M cIntyre,D.R.;Burstein,G.T.;Vossen,A.J.Power Sources 2002,107,67.doi:10.1016/S0378-7753(01)00987-9
(32)Raghuveer,V.;Viswanathan,B.J.Power Sources2005,144, 1.doi:10.1016/j.jpowsour.2004.11.033
(33)Hays,C.C.;Manoharan,R.;Goodenough,J.B.J.Power Sources1993,45,291.doi:10.1016/0378-7753(93)80018-K
(34)Dang,D.;Gao,H.L.;Peng,L.J.;Su,Y.L.;Liao,S.J.;Wang,Y. Acta Phys.-Chim.Sin.2011,27,2379.[党 岱,高海丽,彭良进,苏允兰,廖世军,王 晔.物理化学学报,2011,27,2379.] doi:10.3866/PKU.WHXB20110922
(35)A li,L.I.;A li,A.G.A.;Aboul-Fotouh,S.M.;Aboul-Gheit,A. K.Appl.Catal.A-Gen.1999,177,99.doi:10.1016/S0926-860X (98)00248-8
(36)Lafuente,E.;Muñoz,E.;Benito,A.M.;Maser,W.K.;Martínez, M.T.;A lcaide,F.;Ganborena,L.;Cendoya,I.;M iguel,O.; Rodríguez,J.;Urriolabeitia,E.P.;Navarro,R.J.Mater.Res. 2006,21,2841.doi:10.1557/jm r.2006.0355
(37)Reddington,E.;Sapienza,A.;Gurau,B.;Viswanathan,R.; Sarangapani,S.;Smotkin,E.S.;Mallouk,T.E.Science 1998, 280,1735.doi:10.1126/science.280.5370.1735
(38)Oleg,A.P.J.Solid State Electr.2008,12,609.doi:10.1007/ s10008-007-0500-4
(39)Sun,Y.P.;Xing,L.;Scott,K.J.Power Sources2010,195, 1.doi:10.1016/j.jpowsour.2009.07.028
(40)Luo,J.;Njoki,P.N.;Lin,Y.;Mott,D.;Wang,L.;Zhong,C.J. Langmuir2006,22,2892.doi:10.1021/la0529557
(41)Luo,J.;Maye,M.M.;Kariuki,N.N.;Wang,L.;Njoki,P.;Lin, Y.;Schadt,M.;Naslund,H.R.;Zhong,C.J.Catal.Today 2005, 99,291.doi:10.1016/j.cattod.2004.10.013
(42)Morante-Catacora,T.Y.;Ishikawa,Y.;Cabrera,C.R. J.Electroanal.Chem.2008,621,103.doi:10.1016/j. jelechem.2008.04.029
(43)Neto,A.O.;Dias,R.R.;Tusi,M.M.;Linardi,M.;Spinacé,E. V.J.Power Sources2007,166,87.doi:10.1016/j. jpowsour.2006.12.088
(44)Yi,Q.;Zhang,J.;Chen,A.;Liu,X.;Xu,G.;Zhou,Z.J.Appl. Electrochem.2008,38,695.doi:10.1007/s10800-008-9490-x
(45)Liu,Y.C.;Qiu,X.P.;Huang,Y.Q.;Zhu,W.T.J.Power Sources2002,111,160.doi:10.1016/S0378-7753(02)00298-7
(46)Thomas,J.M.Angew.Chem.Int.Edit.1994,33,913.
(47)Eller,K.;Schwarz,H.Chem.Rev.1991,91,1121.doi:10.1021/ cr00006a002
(48)Kulesza,P.J.;Matczak,M.;Wolkiew icz,A.;Grzybowska,B.; Galkowski,M.;Malik,M.A.;Wieckowski,A.Electrochim. Acta 1999,44,2131.doi:10.1016/S0013-4686(98)00321-1
(49)Gasteiger,H.A.;Markovic,N.;Ross,P.N.;Cairns,E.J. J.Phys.Chem.1993,97,12020.doi:10.1021/j100148a030
(50)Lu,Q.;Li,J.P.Guangdong Chemical Industry 2006,33,8.[陆勤,李俊鹏.广东化工,2006,33,8.],
(51)Zhong,W.;Liu,Y.;Zhang,D.J.Mol.Model.2012,18,3051. doi:10.1007/s00894-011-1318-7
(52)Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;etal.Gaussian 09, Revision A.02;Gaussian Inc.:Wallingford,CT,2009.
(53)Becke,A.D.J.Chem.Phys.1993,98,5648.doi:10.1063/ 1.464913
(54)Hay,P.J.;Wadt,W.R.J.Chem.Phys.1985,82,299.doi: 10.1063/1.448975
(55)Hay,P.J.;Wadt,W.R.J.Chem.Phys.1985,82,270.doi: 10.1063/1.448799
(56)Fukui,K.J.Phys.Chem.1970,74,4161.doi:10.1021/ j100717a029
(57)Neese,F.WIREsComput.Mol.Sci.2012,2,73.doi:10.1002/ w cms.81
(58)Neese,F.J.Am.Chem.Soc 2006,128,10213.doi:10.1021/ ja061798a
(59)Sun,X.L.;Huang,X.R.;Li,J.L.;Huo,R.P.;Sun,C.C. J.Phys.Chem.A 2012,116,1475.doi:10.1021/jp2120302
(60)Sun,X.L.;Geng,C.Y.;Huo,R.P.;Ryde,U.;Bu,Y.X.;Li,J.L. J.Phys.Chem.B 2014,118,1493.
(61)Sun,X.H.;Sun,X.L.;Geng,C.Y.;Zhao,H.T.;Li,J.L. J.Phys.Chem.A 2014,118,7146.doi:10.1021/jp505662x
(62)Huo,R.P.;Zhang,X.;Huang,X.R.;Li,J.L.;Sun,C.C.J.Mol. Model.2013,19,1009.doi:10.1007/s00894-012-1616-8
(63)Sun,X.L.;Li,J.L.;Huang,X.R.;Sun,C.C.Acta Chim.Sin. 2012,70,1245.[孙小丽,李吉来,黄旭日,孙家钟.化学学报,2012,70,1245.]doi:10.6023/A1201134
(64)Huo,R.P.;Zhang,X.;Huang,X.R.;Li,J.L.;Sun,C.C. J.Phys.Chem.A 2011,115,3576.doi:10.1021/jp200231n
(65)Pettersen,E.F.;Goddard,T.D.;Huang,C.C.;Couch,G.S.; Greenblatt,D.M.;Meng,E.C.;Ferrin,T.E.J.Comput.Chem. 2004,25,1605.
(66)Zhang,X.;Schwarz,H.Theor.Chem.Acc.2011,129,389.doi: 10.1007/s00214-010-0861-0
(67)Li,J.L.;Mata,R.A.;Ryde,U.J.Chem.Theory Comput.2013, 9,1799.doi:10.1021/ct301094r
(68)Zhang,X.;Schwarz,H.Chem.-Eur.J.2010,16,5882.doi: 10.1002/chem.201000567
(69)Li,J.L.;Ryde,U.Inorg.Chem.2014,53,11913.doi:10.1021/ ic5010837
(70)Li,J.L.;González-Navarrete,P.;Schlangen,M.;Schwarz,H. Chem.-Eur.J.2015,21,7780.doi:10.1002/chem.201500715
(71)Greeley,J.;Mavrikakis,M.J.Am.Chem.Soc.2002,124, 7193.doi:10.1021/ja017818k
(72)Greeley,J.;Mavrikakis,M.J.Am.Chem.Soc.2004,126, 3910.doi:10.1021/ja037700z
Theo retical Study o fMethano lC―H and O―H Bond Activation by PtRu Clusters
ZHAO Jun-Feng SUN Xiao-Li LIJi-Lai*HUANG Xu-Ri*
(Institute ofTheoreticalChemistry,Jilin University,Changchun 130023,P.R.China)
Density functional theory calculations were performed to study themechanism and reactivity of methanoloxidationmediated by PtnRum(n+m=3,n≠0)clusters.The potentialenergy surfaces and pathways of the initialO―H and C―H bond activationswere predicted.The results show that the activation ofmethanol proceeds preferentially along the C―H bond activation pathway.The calculated reactivity orderwas Pt2Ru>Pt3>PtRu2.Frontierm olecular orbitalanalysis showed that the initialC/O―H bond activation is a proton transfer process.The solventeffectwas also investigated.This study w illenable a deeper understanding of C/O―H bond activation and provide new ideas for catalystselection and optim izing conditions formethanolactivation.
Density functional theory;Cluster;Methanol;Reactivity;Proton transfer
O641
icle]
10.3866/PKU.WHXB201504014 www.whxb.pku.edu.cn