活性污泥处理含氰废水毒性及降解机制研究
2015-12-16张利华张海涛
张利华,周 珉,瞿 贤,张海涛
(1.华东理工大学 化工学院,上海 200237;2.上海化学工业区中法水务发展有限公司,上海 201507)
含氰废水是指含有氰基(—C≡N)的工业废水[1]。工业上氰化物的用途非常广泛,既可作为强络合剂用于提金、电镀工艺,又可作为化工原料用于合成橡胶、纤维、染料、炼焦及有机玻璃等工业[2]。氰化物属于剧毒物质,人一次性服用氰化氢的平均致死量为50~60mg,小动物或牲畜的致死量更小[3]。因此,含氰废水的处理尤为重要。
含氰废水处理方法有多种,主要分为化学法、物理法和生物法[4]。常用的化学法有碱性氯氧化法[5]、过氧化氢氧化法[6]、臭氧氧化法[7]等。常用的物理化学法有活性炭吸附氧化法[8]、溶剂萃取法[9]、离子交换法[10]等。化学法和物理法的成本较高,且会产生其他有害物质,而生物法作为一种经济、环保的方法越来越受重视[11]。由于氰化物对普通微生物具有毒性和抑制性,所以生物法处理含氰废水的研究主要集中在微生物的驯化及特殊菌种的培养(如,P.fluorescens,Pseudomonas sp.)方面[12-13],而利用非驯化、非特殊菌种处理含氰废水的研究还鲜有报道。试验利用传统活性污泥针对含氰废水进行耗氧速率、三磷酸腺苷(ATP)的测定和硝化、反硝化毒性的研究,并初步研究生物法降解废水中氰化物的机制。
1 试验部分
1.1 试验材料与仪器
水样:取自某污水处理厂。
污泥:取自某污水处理厂缺氧池和曝气池。由于未来该污水处理厂会处理含氰废水,而废水中氰化物的最高浓度预计会达到2.0mg/L,因此,试验采用该污水处理厂废水水样中添加不同量的氰化钠固体配置成的模拟废水(氰化钠浓度为0~2.0mg/L)。
试剂:氰化钠,分析纯,国药集团化学试剂有限公司。
BI2000电解呼吸仪(Bioscience,Inc.),FOSS 8400凯氏定氮仪(福斯),DR5000分光光度计(HACH),YSI5100 溶解氧仪(Xylem),pHS-3C型pH计(上海雷磁仪器厂),SKY-8860-2摇床(苏坤),DL-4C型低速大容量夺冠离心机(上海安亭科学仪器厂),MS204S电子天平(Mettler Toledo),Lumitester c-100ATP 检 测 仪 (LuminUltra)。
1.2 试验方法
总氰化物的测定采用硝酸银滴定法(GB7486—1987),ATP的测定采用荧光素酶法,凯氏氮的测定参照硒催化矿化测定方法(ISO5663—1984),硝酸盐氮的测定采用变色酸法(HACH 10020),亚硝酸盐氮的测定采用分子吸收分光光度法(GB/T 7493),耗氧速率的测定采用活性污泥耗氧速率抑制测定标准方法(ISO/DIS 8192),硝化毒性和反硝化毒性的测定采用评估活性污泥微生物硝化、反硝化抑制毒性测定的欧洲标准方法(ISO 9509),生物可处理性评估测试方法参考赞恩-惠伦斯试验(ISO9888:1999(E))。
2 试验结果与讨论
2.1 耗氧速率的测定
在25℃、不同氰化物质量浓度条件下,通过测定微生物的耗氧速率考察氰化物质量浓度对微生物的毒性作用,结果见表1。理论上,废水中若有易于生物降解的物质,则微生物新陈代谢速度加快,从而耗氧速率增大;相反,当废水中加入有毒物质后,微生物的新陈代谢受到抑制,则耗氧速率会降低。
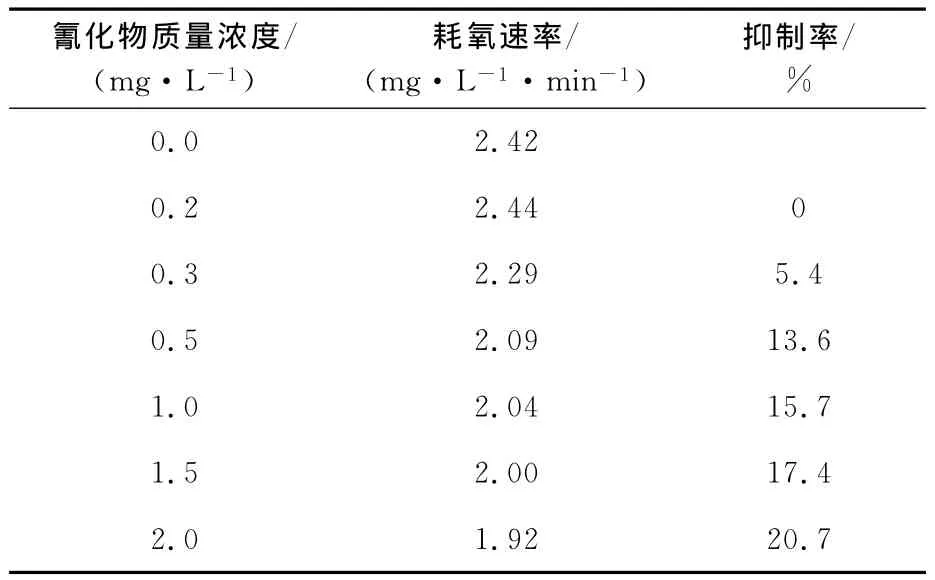
表1 耗氧速率测定结果
由表1看出:随氰化物质量浓度增高,微生物代谢耗氧速率降低;水样中没有氰化物时,微生物耗氧速率为2.42mg/(L·min);水样中氰化物质量浓度为2.0mg/L时,微生物耗氧速率下降20.7%。由此可见,氰化物对微生物的新陈代谢过程有一定抑制作用,但即使氰化物质量浓度达到2.0mg/L,也不会完全抑制微生物活动,因此,利用传统生物法处理含氰废水是可行的。文献[14]报道,生物法能处理氰化物质量浓度高达20 mg/L的含氰废水,根据不同的工艺参数,氰化物去除率可达78%~100%。
2.2 ATP的测定
ATP是指三磷酸腺苷,是生物体内能量代谢的重要产物和细胞代谢可利用能量的携带者,其参与生物体内蛋白质、脂肪的生化合成、吸收等反应。ATP含量可直接反映微生物的活性。试验采用荧光素酶法,在25℃下测定不同氰化物质量浓度下样品中死亡细胞的ATP及活细胞和死亡细胞的总ATP,根据它们的比值得到微生物威胁指数(BSI指数),以此来评估污泥中微生物的活性。BSI指数越高,微生物活性越低。氰化物质量浓度对BSI指数的影响试验结果如图1所示。
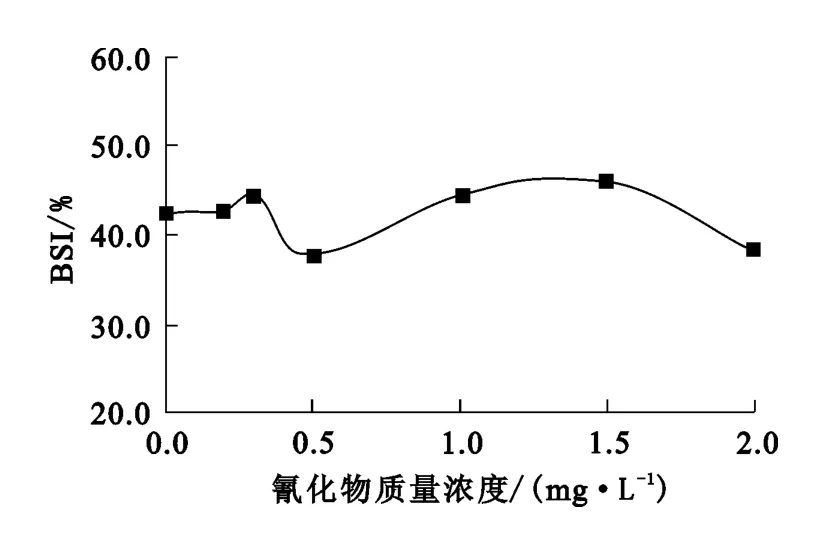
图1 氰化物质量浓度对BSI指数的影响
从图1看出:废水中不含氰化物(0~2.0 mg/L)时,BSI指数为42.6%;废水中加入氰化物后,BSI指数最大为45.9%,无明显变化。说明微生物在氰化物质量浓度为0~2.0mg/L范围内仍具有活性。与耗氧速率测定结果相比较,可以推断,当氰化物质量浓度为0~2.0mg/L时,微生物的新陈代谢受到抑制,表现为耗氧速率降低,但是该浓度范围并没有达到微生物的致死浓度,表现为BSI指数波动平缓。
2.3 硝化毒性测定
硝化毒性的测定是在25℃、不同氰化物质量浓度条件下,通过测定并比较反应初始和24h后水样中氮类物质(如氨氮、凯氏氮等)的转化量,从而得出不同氰化物质量浓度对微生物的抑制率。试验选用凯氏氮作为测试指标,因为所用水样中有机氮的质量浓度较高(10~20mg/L)。在硝化过程中,有机氮会发生氨化反应而转化为氨氮,进而转化为亚硝酸盐氮、硝酸盐氮。氰化物质量浓度对微生物抑制率的影响试验结果如图2所示。可以看出:当水样中氰化物质量浓度为0.2mg/L时,其对硝化过程的抑制率为2.1%;随氰化物质量浓度增大,硝化作用抑制率升高,氰化物质量浓度为1.0mg/L时,抑制率达54.4%;氰化物质量浓度为2.0mg/L时,硝化过程已被完全抑制。试验结果与Y.M.Kim等[15]所得试验结果相似:氰化物质量浓度为0.1mg/L时对硝化过程无抑制;浓度增大,对硝化过程的抑制作用相应增强。
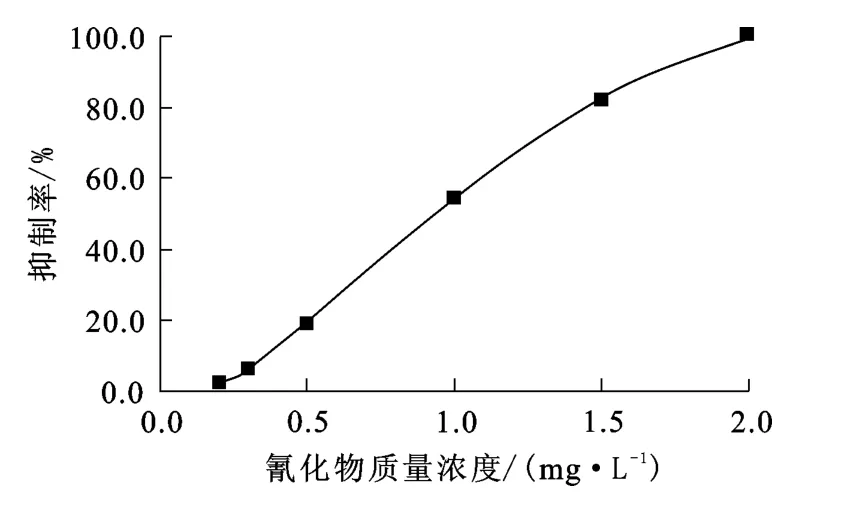
图2 氰化物质量浓度对微生物抑制率的影响
2.4 反硝化毒性测试
微生物反硝化过程分为2步:第1步是从硝酸盐氮转化为亚硝酸盐氮,第2步是从亚硝酸盐氮转化为氮气。反硝化毒性测试是考察在25℃、不同氰化物质量浓度条件下,反应初始和24h后水样中的硝酸盐氮、亚硝酸盐氮的转化量,从而得出不同质量浓度氰化物对微生物反硝化过程抑制率的影响,试验结果如图3所示。
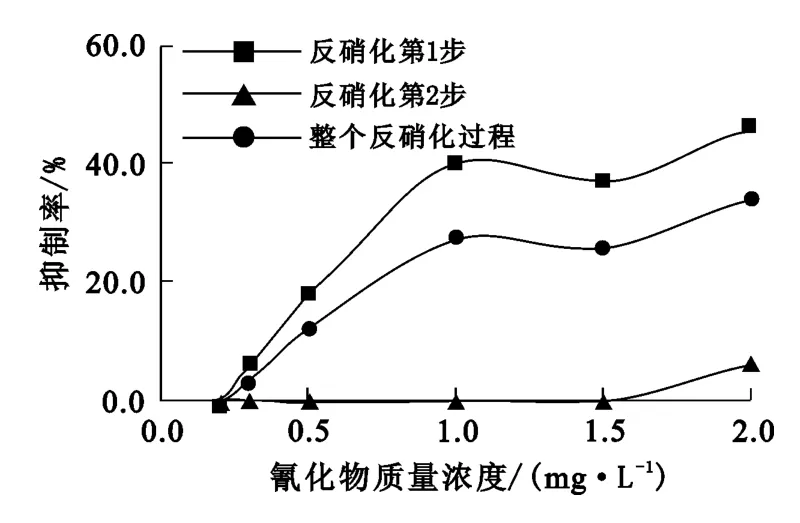
图3 氰化物质量浓度对微生物反硝化过程抑制率的影响
从图3看出:氰化物对反硝化过程第1步有较大抑制作用,氰化物质量浓度从0.2mg/L增大至2.0mg/L,抑制率从0%提高至45.9%;氰化物对反硝化过程第2步无显著抑制作用,在试验范围内,最大抑制率仅为7%。因此,氰化物在反硝化过程中的抑制主要是作用于反硝化过程的第1步,即硝酸盐氮转化为亚硝酸盐氮。这与文献[16-17]的结果相似。
2.5 含氰废水生物降解机制分析
试验在电解呼吸仪(如图4所示)中进行。该装置通过内置供氧系统维持微生物正常新陈代谢活动,无任何外源供氧。与传统赞恩-惠伦斯试验所用反应装置相比,该装置避免了废水水样因挥发而导致的COD下降,可以仅考察废水的生物降解特性。控制温度为25℃,采用模拟含氰废水(纯水+氰化钠固体+营养液),通过测定并比较氰化物的转化量推断氰化物的好氧生物降解性,通过测定并比较氰化物、氨氮、硝酸盐氮和亚硝酸盐氮的转化量,初步分析生物降解氰化物的反应机制。试验结果如图5~7所示。
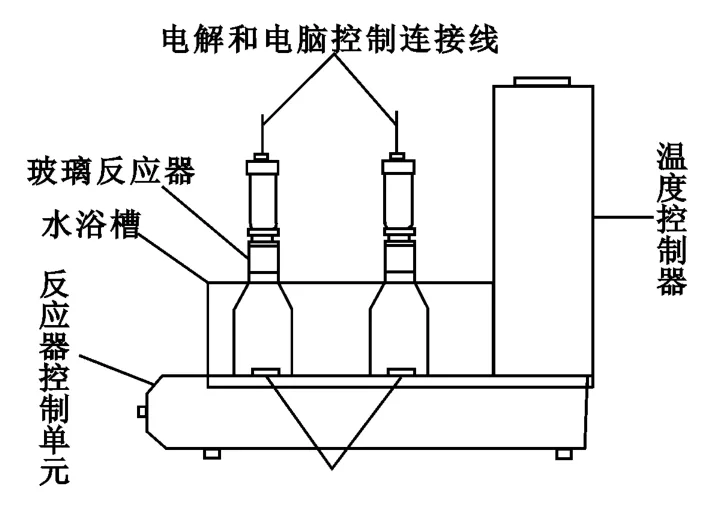
图4 电解呼吸仪
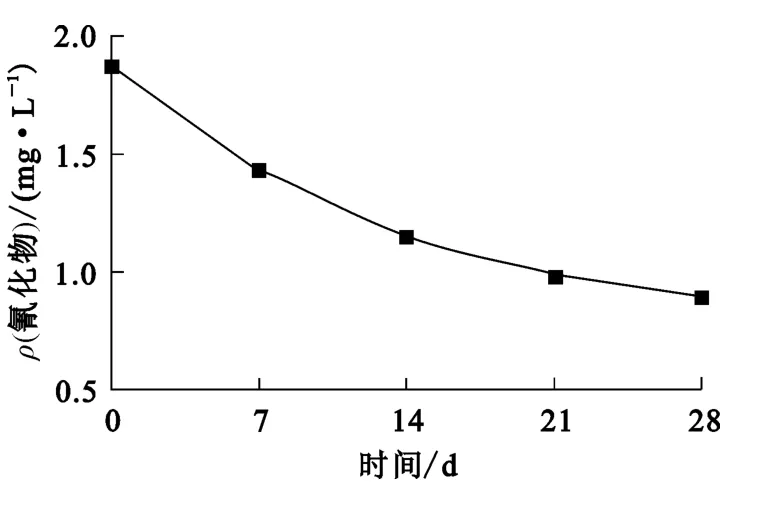
图5 氰化物浓度随反应时间的变化曲线
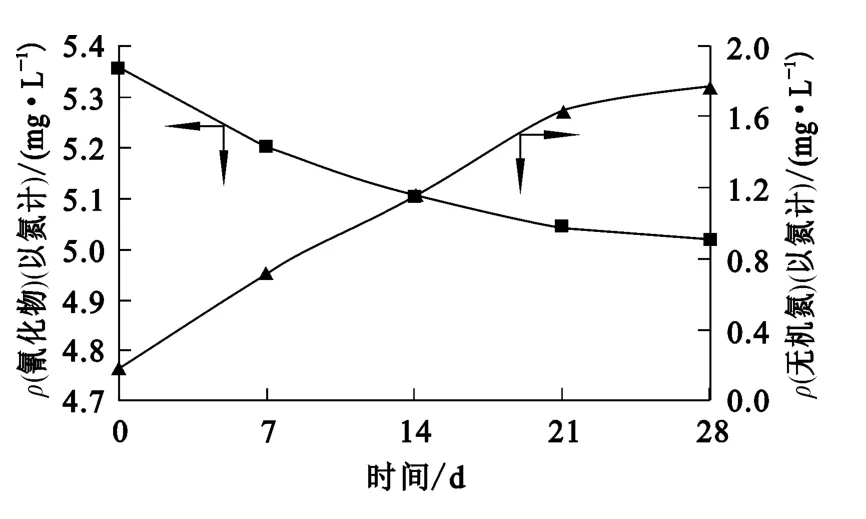
图6 氰化物和无机氮的变化
如图5看出,经过28d,废水水样中氰化物去除率为51.6%,表明活性污泥在氰化物质量浓度为1.87mg/L条件下仍能将部分氰化物降解,这与耗氧速率及ATP测定结果一致,即氰化物质量浓度在0~2.0mg/L范围内,微生物新陈代谢过程受到抑制,但尚未致死,微生物仍有能力进行生物降解。
由图6看出:经过28d的生物降解,废水水样中氰化物(以氮计)质量浓度下降了0.52 mg/L;而水样中无机氮(氨氮、亚硝酸盐氮、硝酸盐氮)质量浓度提高了0.557mg/L。此外,废水经过7d生物降解,COD由初始时的976mg/L降低至32mg/L。由于废水中无其他外源氮存在,因此可以初步推断,微生物降解氰化物是通过将氰根转化为无机氮形式进行的。
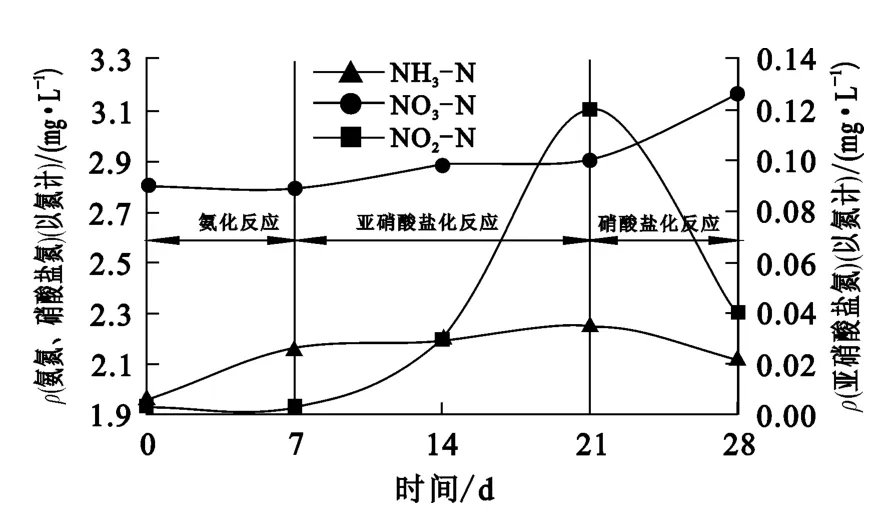
图7 氮浓度随反应时间的变化曲线
由图7看出,在0~7d内,水样中氨氮质量浓度有所增大,亚硝酸盐氮及硝酸盐氮质量浓度无显著变化。结果表明,在降解过程中,由于反应初期废水中有机物含量较高,而氨化细菌为典型异养型微生物,因此其成为优势菌种,能将部分氰化物转化为氨氮;但是由于废水水样中氰化物浓度仍大于1.0mg/L,根据硝化毒性试验结果,硝化过程受到较大抑制,无法将氨氮进一步转化为亚硝酸盐氮和硝酸盐氮。在第8~21d,水样中氨氮浓度继续升高,亚硝酸盐浓度也迅速升高,而硝酸盐氮浓度维持不变,这表明,从第8d开始,由于水样中氰化物浓度降低及微生物对水样适应性增强,亚硝酸盐细菌开始将部分氨氮转化为亚硝酸盐;但是硝酸盐细菌仍受到抑制,从第21~28 d,水样中氨氮及亚硝酸盐氮浓度持续下降,而硝酸盐氮浓度显著上升:这表明,硝酸盐细菌成为优势菌种,并将亚硝酸盐氮转化为硝酸盐氮。从第8~28d天,由于废水水样中的有机物含量较低ρ(COD)<32mg/L),自养型微生物(如硝化细菌)渐渐成为优势菌种,因此硝化过程得以进行。
3 结论
氰化物对微生物的新陈代谢有一定抑制作用,但在0~2.0mg/L范围内不会致死。氰化物质量浓度为2.0mg/L时会完全抑制硝化过程。氰化物质量浓度为2.0mg/L的含氰废水不能直接用活性污泥进行处理。氰化物对微生物反硝化过程的抑制主要是在反硝化的第1步,即硝酸盐氮转化为亚硝酸盐氮。好氧生物降解氰化物的机制为氰化物转化为氨氮,并最终转化为亚硝酸盐氮、硝酸盐氮。
[1]薛文平,薛福德,姜莉莉.含氰废水处理方法的进展与评述[J].黄金,2008,29(4):45-50.
[2]邱廷省,郝志伟,成先雄.含氰废水处理技术与展望[J].江西冶金,2002,22(3):537-539.
[3]卢宜源.贵金属冶金学[M].长沙:中南工业大学出版社,1994:165-167.
[4]陈华进.硫酸亚铁法处理高浓度含氰废水[J].工业水处理,2009,29(10):86-88.
[5]熊如意,乐美承.碱性氯化法处理选矿含氰废水[J].污染防治技术,1998,13(3):18-20.
[6]陈民友,袁玲.采用过氧化氢氧化法处理酸性含氰废水技术研究[J].黄金,1998,19(3):47-51.
[7]李玉峰.臭氧氧化法处理金矿含氰废水的试验研究[J].辽宁城乡环境科技,1999,19(3):16-27.
[8]张明祖,刘建.活性炭负载离子改性及其去除水中氰离子的研究[J].黄金,2008,29(6):51-54.
[9]杨明德,王峻峰,公锡泰.烷基叔胺萃取处理氰化浸金贫液的研究.Ⅰ:萃取体系的选择与工艺实验[J].有色金属,2000,52 (3):61-65.
[10]Osathaphan K,Bconpitak T,Laopirojana T,et al.Removal of Cyanide and Zinc-cyanide Complex by An Ion-exchange Process[J].Water Air Soil Pollution,2008,194(1/4):179-183.
[11]Dash R R,Gaur A,Balomajumder C.Cyanide in Industrial Wastewaters and Its Removal:A Review on Biotreatment[J].Journal of Hazardous Materials,2009,163(1):1-11.
[12]Gurbuz F,Ciftci H,Akcil A.Biodegradation of Cyanide Containing Effluents by Scenedesmusobliquus[J].Journal of Hazardous Materials,2009,162(1):74-79.
[13]Huertasa M J,Sáez L P,Roldán M D,et al.Alkaline Cyanide Degradation by Pseudomonas Pseudoalcaligenes CECT5344in A Batch Reactor:Influence of pH[J].Jour-nal of Hazardous Materials,2010,179(1/3):72-78.
[14]White D M,Pilon T A,Woolard C.Biological Treatment of Canide Containing Wastewater[J].Water Research,2000,34(7):2105-2109.
[15]Kim Y M,Park D,Lee D S,et al.Inhibitory Effects of Toxic Compounds on Nitrification Process for Cokes Wastewater Treatment[J].Journal of Hazardous Materials,2008,152(3):915-921.
[16]Kim Y M,Lee D S,Park C,et al.Effects of Free Cyanide on Microbial Communities and Biological Carbon and Nitrogen Removal Performance in the Industrial Activated Sludge Process[J].Water Research,2011,45(3):1267-1279.
[17]Kim Y K,Chou H U,Lee D S,et al.Comparative Study of Free Cyanide Inhibition on Nitrification and Denitrification in Batch and Continuous Flow Systems[J].Desalination,2011,279(1/3):439-444.
