不同龋敏感青少年口腔细菌多样性分析
2015-12-16张洋洋何金枝徐欣周学东
张洋洋 何金枝 徐欣 周学东
1.新疆医科大学附属口腔医院牙体牙髓科,乌鲁木齐 830054;2.口腔疾病研究国家重点实验室 华西口腔医院(四川大学),成都 610041
龋病是口腔细菌、可发酵的碳水化合物和易感牙齿经长时间复杂的相互作用而发生局部破坏的感染性疾病[1]。近年来多个国家的流行病学数据表明龋病患病率正显著增加,人们面临一场严峻的公共健康危机[2],因此龋病的防治应引起全世界更多的重视。目前关于口腔健康研究中的对象多数是儿童,而关于青少年口腔健康的数据非常少[3]。我国第二次全国口腔健康流行病学抽样调查报告显示,12岁青少年的患龋率为48.32%;15岁青少年的患龋率为55.70%;18岁青少年的患龋率男性为55.32%,女性为60.11%[4]。我国第三次全国口腔健康调查报告显示,12岁青少年的患龋率为28.90%,与前次调查结果相比,虽然情况有所好转,但口腔健康状况并没有从根本上得到改善。因此,关注青少年口腔健康、了解青少年龋病状态下口腔细菌多样性的变化对于青少年这一特定群体龋病的防治是非常必要的。
龋病发生的环境因素非常复杂,目前已经达成的医学共识就是微生物因素:口腔微生物代谢碳水化合物产酸,导致釉质表面的脱矿和再矿化过程失衡[5]。研究口腔内定植细菌种类及其致病潜能是防治龋病的基础[6]。对不同疾病状态下口腔微生物组成多样性的认识,能够更好地揭示疾病的发病机制[7]。唾液细菌可作为口腔微生物的“指纹”,指示口腔生境内微生物菌群结构的变化[8],且唾液易于采集、保存和运送,并可减少患者的焦虑和不适。
环境样本中超过99%的细菌无法在实验室进行培养[9]。变性梯度凝胶电泳(denaturing gradient gel electrophoresis,DGGE)结合聚合酶链反应(polymerase chain reaction,PCR)不仅能够检测可培养细菌,还能够检测出一些稀有的、难培养的细菌,进而更有效地评估微生物多样性和比较不同环境微生物群落结构的差别。近年来,DGGE技术广泛应用于微生物多样性研究中,涉及湿地[10]、草地[11]和食品[12]等各个领域,在口腔微生物多样性研究中的应用也日趋增多,如龋病[13-14]、牙髓及根尖周病[15-17]和牙周病[18]患者的口腔微生物多样性研究等。本实验以唾液为样本,采用PCR-DGGE指纹图谱技术对不同龋状况青少年的唾液样本进行研究,通过分析不同龋状况青少年的唾液细菌多样性差异,初步了解青少年龋病状态下口腔微生物组成的群体性改变,为针对龋病特定人群进行有效防治提供理论依据。
1 材料和方法
1.1 研究对象的选择
2011年12月—2012年6月从四川大学华西口腔医院初诊人群中随机选取20名12~18岁青少年,分为两组:无龋组和高龋组。无龋组:女孩6名,男孩4名,平均年龄15岁;纳入标准:诊断为无龋的青少年(诊断标准:DMFT=0[19])。高龋组:女孩6名,男孩4名,平均年龄15岁;纳入标准:诊断为高龋的初诊青少年(诊断标准:DMFT≥6[19]);排除标准:有全身性疾病或除龋病以外的任何口腔疾病,或近6个月内使用过抗生素等药物。本研究经过四川大学华西口腔医院伦理委员会批准。研究对象或其监护人在取样前均签署知情同意书。
1.2 唾液样本的采集
使用1.5 mL无菌离心管直接收集1 mL非刺激性唾液。指导受试者停止吞咽动作1 min后让唾液自行流入1.5 mL无菌离心管,重复这一操作过程直至收集到1 mL唾液。样本冰浴下2 h内转送至实验室,-80 ℃冰箱保存备用。
1.3 细菌全基因组DNA提取和PCR扩增
所有样本室温解冻,涡旋3 min使样品充分分散至TE 缓冲液内,弃棉签留菌悬液备用。使用QIAamp DNA Micro Kit(Qiagen公司,美国)提取细菌全基因组DNA作为模板,使用通用细菌16S rRNA引物Bac1和Bac2为引物[20],引物序列如下。Bac1:5'-ACTACGTGCCAGCAGCC-3',Bac2:5'-GGACTACCAGGGTATCTAATCC-3'。为了防止DNA双链完全解离,需要在Bac1 5'端连接40 bp的“GC夹[21]”,序列如下。Bac1:5'-CGCCCGCCGCGCCCCGCGCCCGTCCCGCCGCCCCCGCCCGACTACGTGCCAGCAGCC-3',Bac2:5'-GGACTACCAGGGTATCTAATCC-3',扩增产物长度为300 bp。每50 μlPCR反应体系包含:纯化基因组 DNA(10 ng·μL-1)2 μL、上游及下游PCR引物(25 μmol·L-1)各1 μL、无酶水21 μL、DreamTaqTMPCR Master Mix(2×)(Fermentas公司,立陶宛)25 μL。使用Gradient PCR Thermal Cycle (Eppendorf公司,德国)进行聚合。循环条件为:94 ℃预变性3 min;94 ℃变性1 min,56 ℃退火1 min,72 ℃延伸2 min,30个循环;最后72 ℃延伸5 min。取PCR扩增产物5 μL进行1%琼脂糖凝胶电泳验证,Goldview Ⅰ型核酸燃料染色后置长波紫外光下观察,采用凝胶成像系统(Bio-Rad公司,美国)采集图像。
1.4 DGGE分析
使用Bio-Rad D-Code系统(Bio-Rad公司,美国)对PCR扩增产物进行DGGE。丙烯酰胺凝胶质量分数为8%,变性剂浓度梯度质量分数为40%~60%(100 %变性剂包含体积分数为40%的甲酰胺与7 mol·L-1的尿素),变性胶上表面加入5 mL无变性作用的积层凝胶。60 V恒定电压、58 ℃条件下,在1×TAE缓冲液(pH=8.5)中电泳16 h。电泳完成后,0.5 μg·mL-1溴化乙锭染色15 min,1×TAE缓冲液漂洗去除多余染料,凝胶成像系统采集并记录DGGE 凝胶的数码图像。
1.5 DGGE凝胶微生物图谱分析
使用Quantity One软件(Bio-Rad公司,美国)标准化DGGE凝胶图像用于分析条带模式,得到的最终参数包括检测到的条带数、条带分布的频率等。
1.5.1 Shannon-Wiener's指数 Shannon-Wiener's指数(H')代表微生物群落多样性。H'的计算是基于DGGE胶条带的位置和条带的强度,计算公式为:H'=-∑PilnPi,条带的强度(Pi)由凝胶分析软件Quantity One分析后得到的波峰面积来表示,即Pi=ni/N,其中ni为峰面积,N为所有峰的总面积。
1.5.2 非加权组平均法(unweighted pair-group method with arithmetic mean,UPGMA)聚类分析 使用UPGMA进行聚类分析构建系统进化树。
1.6 统计学分析
采用SPSS 19.0软件分析数据,组间条带数和Shannon-Wiener's指数的比较采用独立样本Mann-WhitneyU检验,检验效能α=0.05。
2 结果
2.1 PCR扩增结果
PCR扩增唾液菌群的16S rDNA,所有样本均得到预期长度约为300 bp的扩增产物(图1);扩增产物无明显拖尾及引物二聚体形成。
图1 PCR产物琼脂糖电泳Fig 1 Agarose gel electrophoresis of PCR products
2.2 DGGE图谱总体特征
图2为高龋和无龋青少年组唾液菌群的PCRDGGE图谱。图中每一泳道代表一个研究对象唾液菌群,不同位置的条带代表不同的优势细菌,泳道的条带数代表该样本中的优势菌种的数量。图2表明:1)个体间唾液细菌中共享的优势菌群数量较多,图谱表现为相同位置的条带在各个体中均有出现;2)唾液细菌的组成同时也具有个体的差异性,图谱表现为未见到完全相同的泳道出现。
2.3 条带数比较
10个高龋青少年及10个无龋青少年唾液样本基因组DNA的DGGE图谱经过标准化后,共得到76个不同的条带,各泳道条带数目介于22~38之间,平均条带数为29.9±4.3。高龋组和无龋组的条带数分别为27.3±3.4和32.5±3.7,差异具有统计学意义(P=0.008)。
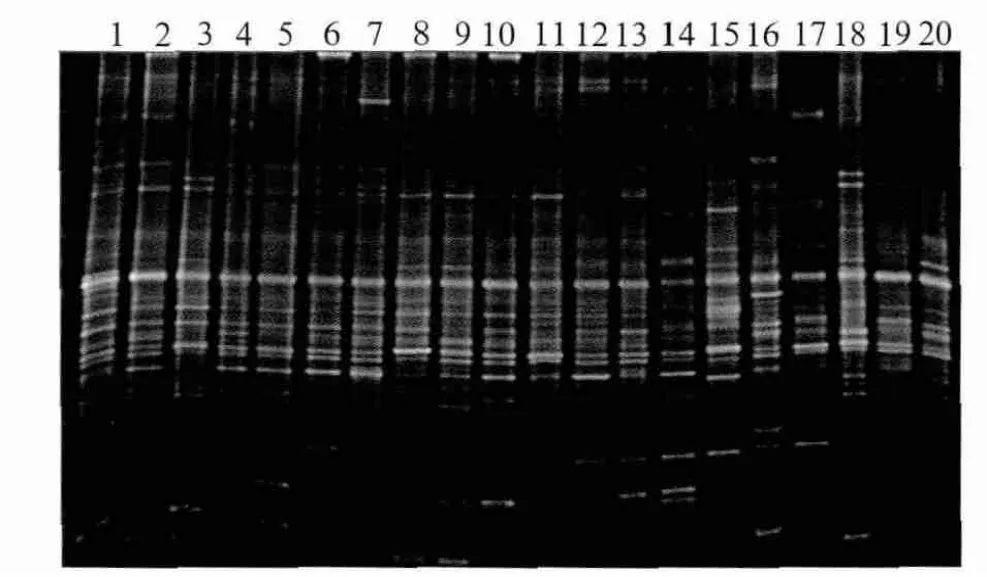
图2 唾液菌群PCR-DGGE图谱Fig 2 PCR-DGGE patterns from salivary microbial
2.4 Shannon-Wiener's指数比较
高龋组和无龋组的Shannon-Wiener's指数分别为2.5±0.2和2.6±0.2,二者之间的差异无统计学意义(P=0.405)。
2.5 UPGMA聚类分析
UAPGM聚类分析结果(图3)表明:3、6、8、9和10号样本在树图中聚成一簇,1、2、4和5号样本在树图中聚成一簇,样本都来自无龋组;16和18号样本在树图中聚成一簇,11、15和17号样本在树图中聚成一簇,19和20号样本在树图中聚成一簇,7、12和13号样本在树图中聚成一簇,除了7号样本属于无龋组以外,其余样本均来自高龋组。同组内大部分样本聚类位置相近,群落结构表现出较高的相似性;不同组的大部分样本未聚类在一起,群落结构呈现出一定的差异。该结果提示唾液细菌的群落结构随着龋病严重程度的增加发生群体性变化,但是每个节点处代表相似性的数字并不是很大,这对聚类分析结果的可信度有一定的影响。
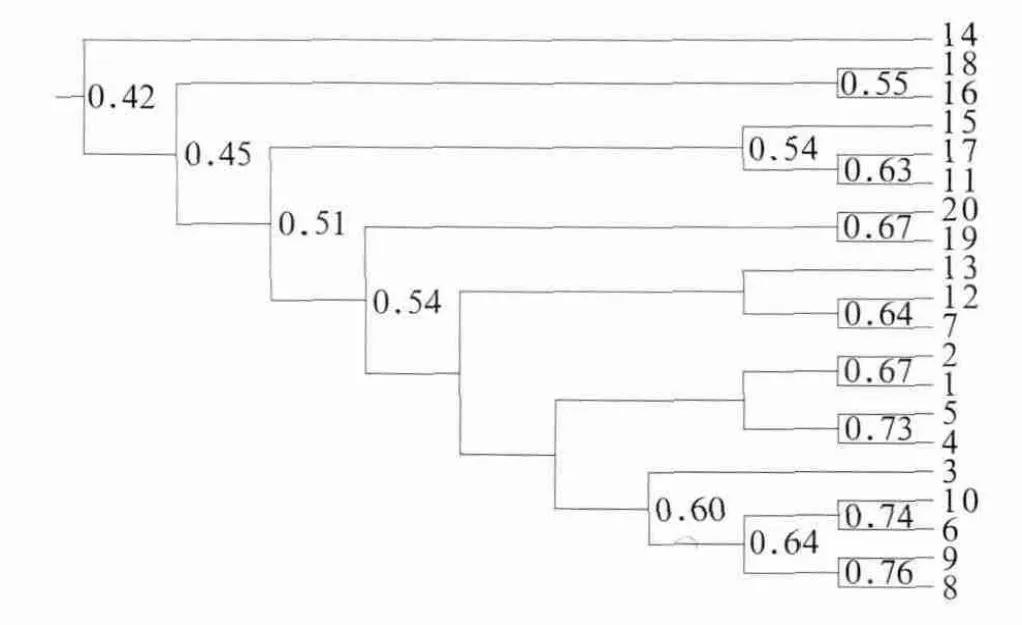
图3 UPGAM聚类分析Fig 3 Cluster analysis with UPGAM
3 讨论
本研究选择12~18岁青少年为研究对象可以更好地了解恒牙龋对青少年的影响。本研究结果表明:1)颊黏膜菌群的组成具有个体差异性;2)高龋青少年唾液样本中检测到的条带数为27.3±3.4,无龋青少年唾液样本中检测到的条带数为32.5±3.7,高龋青少年唾液中的优势菌群数量少于无龋青少年唾液中的优势菌群数量,差异具有统计学意义(P=0.008)。3)高龋青少年唾液细菌多样性的Shannon-Wiener's指数为2.5±0.2,无龋青少年唾液细菌多样性的Shannon-Wiener's指数为2.6±0.2,二者之间无统计学差异(P=0.405)。4)群落结构聚类分析表明,同组内大部分样本聚类位置相近,群落结构表现出较高的相似性;不同组的大部分样本未聚类在一起,群落结构呈现出一定的差异。
从本研究结果可以看出,高龋青少年唾液细菌的多样性低于无龋青少年,唾液细菌的多样性在龋病发展过程中有减少的趋势。产生这一结果的原因可能是[22-23]:1)在龋病发展过程中,某些细菌被淘汰,导致患龋者菌种数降低;2)患龋者拥有更多的产酸和耐酸菌,抑制了其他菌种;3)龋损部位更有利于龋相关菌的繁殖,导致龋相关菌种类数量上升,而其他菌种类数量降低。
为了探索龋病发生、发展过程中微生物群落结构发生的变化,学者们使用不同的方法对不同的口腔微生物样本进行了研究。Li等[24]于2005年首次将PCR-DGGE技术应用于龋病相关微生物研究,发现无龋者唾液细菌种类多于龋活跃者唾液细菌种类,这与本研究的结果相同。一些学者[25-26]也得出了相同的结论。但是也有学者[14,19]得出了不同的结论,认为无龋个体的口腔微生物多样性与有龋个体差异不大。
本研究对不同龋状况唾液细菌群落的差异进行了分析,确认了在龋病发展过程中龋病相关微生物群落多样性减少,为青少年龋病相关的微生物学研究提供了参考。由于本研究样本量不够大,其结果只能作为龋病发生、发展过程中微生物群体变化的初步探索,今后还需要扩大样本量、采用更先进的技术对微生物群体的变化过程进行深入研究。
[1]Selwitz RH, Ismail AI, Pitts NB. Dental caries[J]. Lancet,2007, 369(9555):51-59.
[2]Bagramian RA, Garcia-Godoy F, Volpe AR. The global increase in dental caries. A pending public health crisis[J].Am J Dent, 2009, 22(1):3-8.
[3]Pontigo-Loyola AP, Medina-Solis CE, Borges-Yañez SA,et al. Prevalence and severity of dental caries in adolescents aged 12 and 15 living in communities with various fluoride concentrations[J]. J Public Health Dent, 2007, 67(1):8-13.
[4]全国牙病防治指导组. 第二次全国口腔健康流行病学抽样调查[M]. 北京: 人民卫生出版社, 1999:417-424.
[5]Russell RR. Changing concepts in caries microbiology[J].Am J Dent, 2009, 22(5):304-310.
[6]Paster BJ, Olsen I, Aas JA, et al. The breadth of bacterial diversity in the human periodontal pocket and other oral sites[J]. Periodontol 2000, 2006, 42:80-87.
[7]Siqueira JF Jr, Rôças IN. Community as the unit of pathogenicity: an emerging concept as to the microbial pathogenesis of apical periodontitis[J]. Oral Surg Oral Med Oral Pathol Oral Radiol Endod, 2009, 107(6):870-878.
[8]Fábián TK, Fejérdy P, Csermely P. Salivary genomics, transcriptomics and proteomics: the emerging concept of the oral ecosystem and their use in the early diagnosis of cancer and other diseases[J]. Curr Genomics, 2008, 9(1):11-21.
[9]Sharma R, Ranjan R, Kapardar RK, et al. Unculturable bacterial diversity: an untapped resource[J]. Curr Sci, 2005,89(1):72-77.
[10]Faulwetter JL, Gagnon V, Sundberg C, et al. Microbial processes influencing performance of treatment wetlands:a review[J]. Ecol Eng, 2009, 35(6):987-1004.
[11]Borch T, Kretzschmar R, Kappler A, et al. Biogeochemical redox processes and their impact on contaminant dynamics[J]. Environ Sci Technol, 2010, 44(1):15-23.
[12]Ercolini D. PCR-DGGE fingerprinting: novel strategies for detection of microbes in food[J]. J Microbiol Methods, 2004,56(3):297-314.
[13]Li Y, Ge Y, Saxena D, et al. Genetic profiling of the oral microbiota associated with severe early-childhood caries[J].J Clin Microbiol, 2007, 45(1):81-87.
[14]Ling Z, Kong J, Jia P, et al. Analysis of oral microbiota in children with dental caries by PCR-DGGE and barcoded pyrosequencing[J]. Microb Ecol, 2010, 60(3):677-690.
[15]Siqueira JF Jr, Rôças IN, Rosado AS. Application of denaturing gradient gel electrophoresis (DGGE) to the analysis of endodontic infections[J]. J Endod, 2005, 31(11):775-782.
[16]Siqueira JF Jr, Rôças IN, Debelian GJ, et al. Profiling of root canal bacterial communities associated with chronic apical periodontitis from Brazilian and Norwegian subjects[J]. J Endod, 2008, 34(12):1457-1461.
[17]Zoletti GO, Carmo FL, Pereira EM, et al. Comparison of endodontic bacterial community structures in root-canaltreated teeth with or without apical periodontitis[J]. J Med Microbiol, 2010, 59(Pt 11):1360-1364.
[18]Ledder RG, Gilbert P, Huws SA, et al. Molecular analysis of the subgingival microbiota in health and disease[J]. Appl Environ Microbiol, 2007, 73(2):516-523.
[19]Yang F, Zeng X, Ning K, et al. Saliva microbiomes distinguish caries-active from healthy human populations[J]. ISME J, 2012, 6(1):1-10.
[20]Rupf S, Merte K, Eschrich K. Quantification of bacteria in oral samples by competitive polymerase chain reaction[J].J Dent Res, 1999, 78(4):850-856.
[21]Sheffield VC, Cox DR, Lerman LS, et al. Attachment of a 40-base-pair G + C-rich sequence (GC-clamp) to genomic DNA fragments by the polymerase chain reaction results in improved detection of single-base changes[J]. Proc Natl Acad Sci USA, 1989, 86(1):232-236.
[22]Beighton D. The complex oral microflora of high-risk individuals and groups and its role in the caries process[J].Community Dent Oral Epidemiol, 2005, 33(4):248-255.
[23]Becker MR, Paster BJ, Leys EJ, et al. Molecular analysis of bacterial species associated with childhood caries[J]. J Clin Microbiol, 2002, 40(3):1001-1009.
[24]Li Y, Ku CY, Xu J, et al. Survey of oral microbial diversity using PCR-based denaturing gradient gel electrophoresis[J].J Dent Res, 2005, 84(6):559-564.
[25]Jiang W, Jiang Y, Li C, et al. Investigation of supragingival plaque microbiota in different caries status of Chinese preschool children by denaturing gradient gel electrophoresis[J]. Microb Ecol, 2011, 61(2):342-352.
[26]刘怡然, 王伟健, 沈家平, 等. PCR-DGGE检测早期儿童龋治疗前后口腔菌群多样性的初步研究[J]. 口腔生物医学, 2012, 3(4):201-204.
