糖尿病心肌病中微血管病变的研究进展
2015-12-16颜贵英胡松综述毛拥军审校
颜贵英、胡松综述,毛拥军审校
综述
糖尿病心肌病中微血管病变的研究进展
颜贵英、胡松综述,毛拥军审校
高血糖所致内皮功能障碍,激素的产生和释放的改变,平滑肌细胞的代谢变化这三大主要损伤机制,导致小动脉和微血管的损伤,促进糖尿病心肌病的发展。这些因素通过单独地或联合作用引起氧化应激反应、细胞信号和基因转录改变,导致血管收缩和冠脉结构重建,使心肌灌注不足,从而降低了能量状态,导致Ca2+浓度改变、细胞凋亡、心肌收缩力降低等心功能障碍。本文对糖尿病诱发的以心脏微血管病变为特点的心功能障碍的损伤机制进行综述。
糖尿病;微血管病;心功能障碍;代谢障碍;内皮功能紊乱
糖尿病心肌病发病率高、危害性大,是糖尿病患者的主要并发症之一。糖尿病心肌病是糖尿病引起心脏微血管病变和心肌代谢紊乱所致的心肌广泛局灶性坏死,表现为早期心肌顺应性降低和舒张期充盈受阻的舒张功能不全,晚期收缩功能不全[1]。多项证据表明微血管病变在糖尿病患者心功能障碍的发生中发挥着重要作用[2-4]。联合高血压和糖尿病大鼠实验表明,微循环的损害可能导致局灶性坏死和疤痕形成,进而发展成心肌肥厚和心力衰竭[5,6]。微血管病变主要表现为心脏X综合征,微血管功能障碍,非梗阻性冠状动脉疾病,或微血管心绞痛,很多假设都建立在糖尿病患者心肌收缩力降低的基础上[7,8]。本文旨在阐述糖尿病诱导包括小动脉和毛细血管在内的微血管病变的几种机制。
1 糖尿病相关代谢障碍促进微血管病变
糖尿病的主要临床症状高血糖,是胰岛素缺乏和胰岛素抵抗或两者共同作用的结果[9]。胰岛素除了调节糖类和脂类新陈代谢以外还维持细胞的稳态,胰岛素分泌功能异常可能对心脏和平滑肌产生有害影响(图1)。
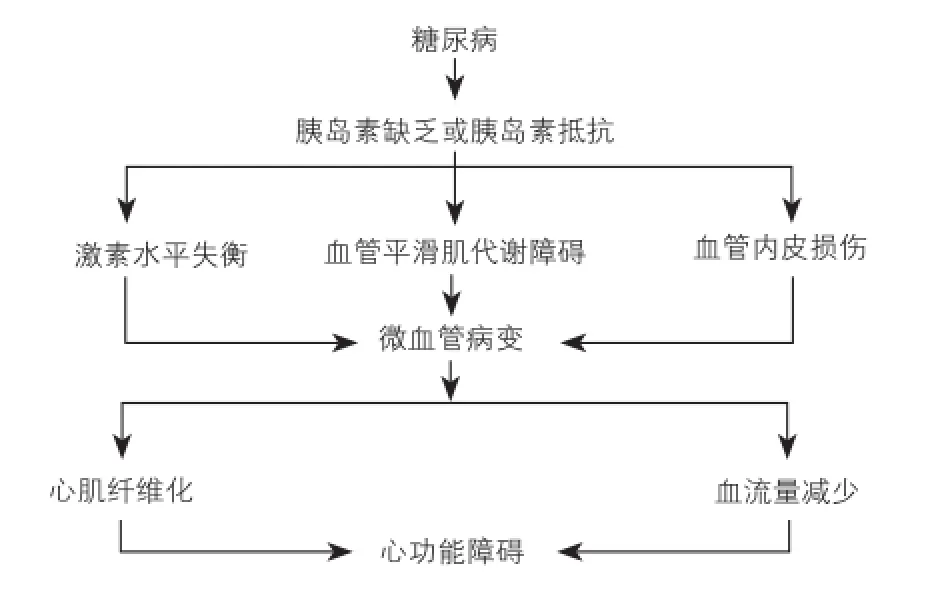
图1 糖尿病诱导心功能障碍的主要机制
糖类、脂肪、游离脂肪酸(FFA)代谢等各种生化改变也提示糖尿病和心血管并发症的联系。在糖尿病患者中,氧化应激是这些代谢障碍的共同特点[8]和被认为可直接或间接地引起内皮功能障碍和脉管系统改变(图2)。
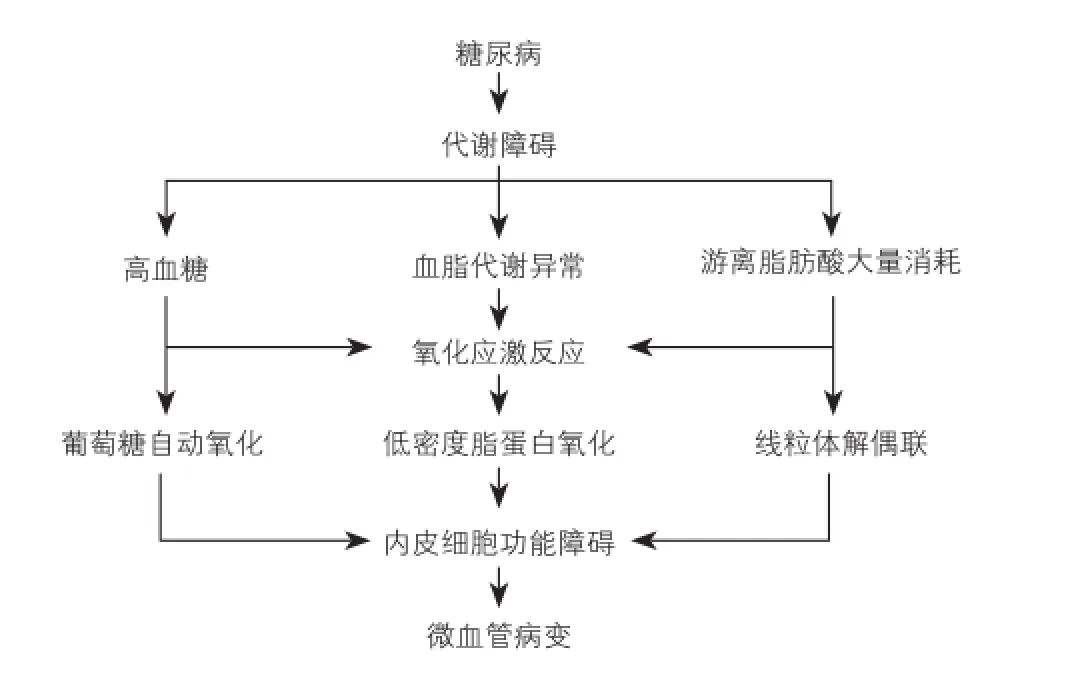
图2 糖尿病引起的各种代谢障碍
1.1 高血糖与糖尿病心肌病微血管病变
高血糖能导致晚期糖基化终产物(AGE)的形成,AGE来源于动脉内皮细胞和心脏的葡萄糖和脂肪酸氧化作用[10,11]。AGE导致了活性氧(ROS)的产生和核因子-κ B(NF-κ B)激活后各种基因转录。NF-κB控制的基因参与炎症、应激、细胞存活、生长因子和增殖[12,13]。一项研究显示,AGE水平和左心室舒张末期内径有直接联系[14]。高血糖降低缺氧诱导因子(HIF-1α)的水平以及内皮前体细胞中内皮一氧化氮合酶(eNOS)的表达[15,16]。这和在糖尿病心肌中HIF-1α[17]的低表达以及由于血管内皮生长因子表达下调和其信号通路[18]所致的冠脉毛细血管密度的减少是一致的。
高血糖可能激活了蛋白激酶C(PKC),通过对胰岛素受体或底物丝氨酸/苏氨酸高度磷酸化来抑制胰岛素的代谢作用导致微血管病。此外,PKC的激活可能改变磷脂酰肌醇3-激酶/蛋白激酶B和丝裂原活化蛋白激酶等其他蛋白激酶的活动,反过来损伤内皮细胞功能和促进血管收缩(图3)[7]。
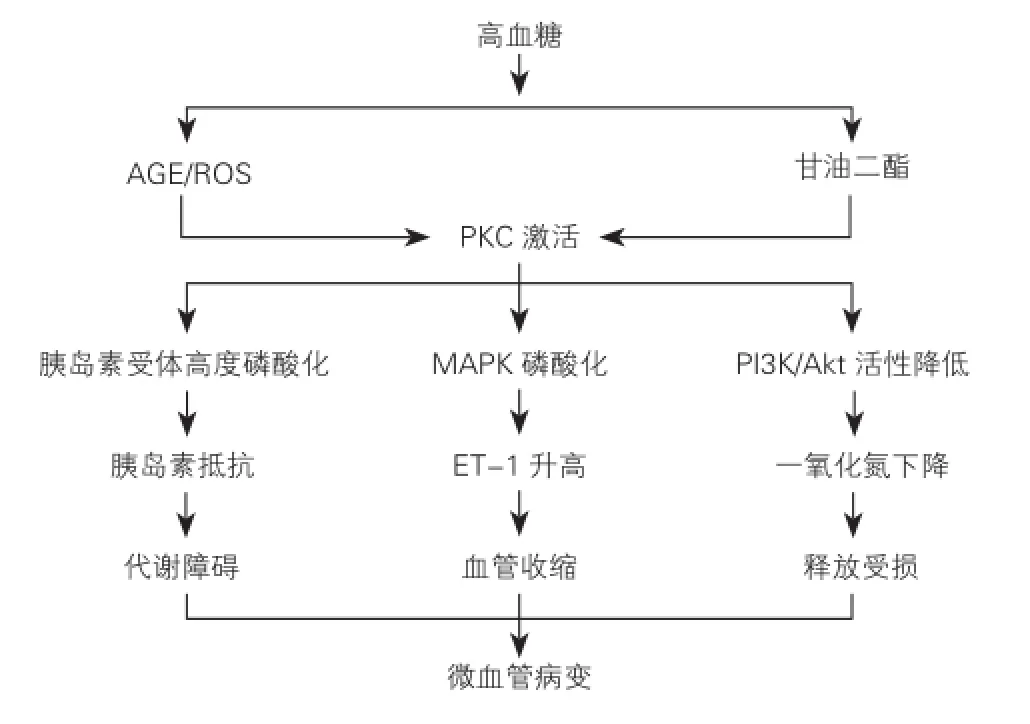
图3 高血糖微血管病变的事件原理图
1.2 血脂代谢异常与糖尿病心肌病微血管病变
糖尿病患者脂类代谢障碍,动脉粥样硬化的单核巨噬细胞和泡沫细胞是内皮细胞功能障碍的关键步骤[19]。有报道指出,单核细胞中激活的PKC可能参与了最初的血管黏附和巨噬细胞的分化[20]。已有证据证明,在糖尿病状态下,酶的抑制作用显著地减弱了巨噬细胞对低密度脂蛋白的摄取[21]。PKC的抑制作用是通过降低细胞黏附因子-1的水平来减弱巨噬细胞活性的[22]。失活的抗动脉粥样硬化酶、内皮型一氧化氮合酶等相互作用致内皮细胞功能障碍。
1.3 游离脂肪酸过度利用与糖尿病心肌病微血管病变
升高的FFA通过分别抑制胰岛素刺激的葡萄糖摄取、糖原合成、胰岛素介导的抑制肝糖分解产生外周和肝的胰岛素抵抗[23]。以FFA升高为主的血脂代谢异常可能导致葡萄糖氧化减少和线粒体解偶联,可能造成心肌高能量储备减少和最终心功能障碍[24,25]。升高的FFA也能调节K+三磷酸腺苷(ATP)通道[26],它的作用为缩短动作电位时程和减少Ca2+内流,以减轻心肌收缩力。升高的FFA的其他有害作用包括心肌脂肪变性和鞘脂类、神经酰胺类的生成诱导凋亡细胞的死亡[24]。已有证据表明,糖尿病患者心肌脂质堆积,出现左心室舒张功能障碍[27]。
2 神经体液系统在糖尿病心肌病微血管病变中的作用
高血糖激活肾素-血管紧张素-醛固酮系统(RAAS)导致血管紧张素(AT)-Ⅱ生成[28],它能通过烟酰胺腺嘌呤二核苷酸/尼克酰胺腺嘌呤二核苷酸磷酸氧化酶产生ROS。循环系统中儿茶酚胺过量导致血管平滑肌Ca2+超载,促进微血管病的进展,发生心肌灌注不足,导致心功能障碍(图4)。此外,这些细胞毒性物质可能直接导致肌纤维膜和肌质网膜的改变,导致细胞内Ca2+超载,发生心功能障碍[29]。
一项研究已报告,缬沙坦通过下调X-盒结合蛋白1表达抑制糖尿病心肌病大鼠心肌细胞凋亡[30]。在从链脲菌素糖尿病大鼠分离的心肌细胞中,AT1受体的表达和AT1受体的密度都增加了[28]。
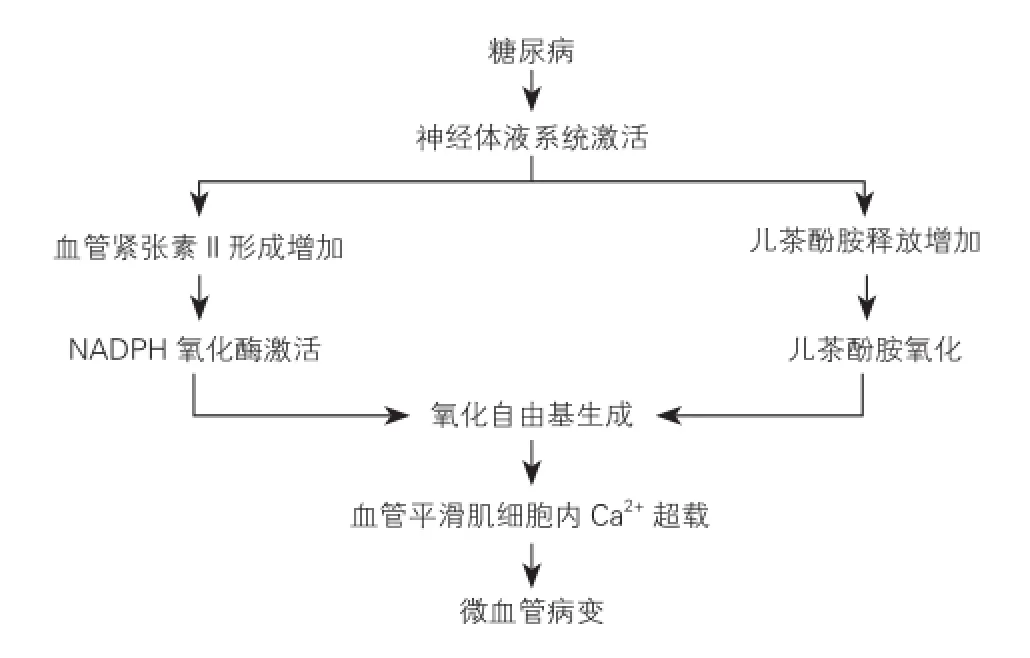
图4 神经体液系统在糖尿病诱导的微血管病中参与血管平滑肌的改变
3 内皮功能障碍在糖尿病心肌病微血管病变中的作用
已有研究证明,内皮细胞功能障碍在糖尿病微血管并发症中起着重要作用[31-33]。由于高血糖,血管扩张剂一氧化氮的生物利用度降低,血管收缩剂内皮素-1水平升高,导致微血管舒张功能障碍促进了微血管病发生[34]。
3.1 内皮素-1与微血管病变
有证据表明,内皮素-1与血栓素A2相互作用调节血管收缩和血小板聚集[35]。此外,内皮素-1促进糖尿病心肌病纤维化发展的另一个机制是由内皮细胞向间叶细胞起源的纤维母细胞的转化[36]。
3.2 一氧化氮与微血管病变
超氧自由基生成细胞毒性氧化剂,过氧化亚硝酸盐[37],可能因使二磷酸核糖腺苷聚合酶活化产生血管功能障碍。糖酵解和柠檬酸循环功能损害,导致ATP的消耗和坏死[38]。过氧化亚硝酸盐介导的作用可能直接诱导小血管收缩(图5)。
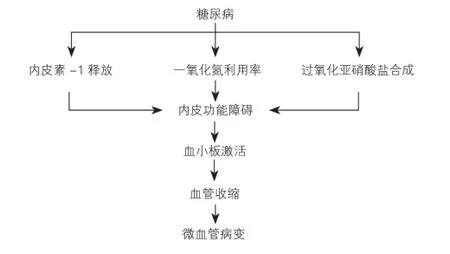
图5 糖尿病内皮功能障碍和血小板活化在微血管病变中的作用
4 结语
综上所述,糖尿病心肌病中胰岛素抵抗或缺乏所致的代谢性障碍和神经体液系统障碍通过协同作用导致微血管病。因此,内皮细胞功能和通透性的改变,平滑肌细胞活性增加,血小板的激活,单核细胞及血小板的黏附可能引起血管收缩,激活纤维化信号通路。对微血管的这些损伤中,氧化应激、PKC的激活、NF-κB水平的升高进一步改变心肌结构和功能。因此,微血管病变在糖尿病心肌病中起着重要作用,可能为糖尿病心肌病的防治策略提供新思路。
参考文献
[1] 冯新星, 陈燕燕. 糖尿病心肌病的研究进展. 中国循环杂志, 2015, 29: 87-89.
[2] Pappachan JM, Varughese GI, Sriraman R, et al. Diabetic cardiomyopathy: Pathophysiology, diagnostic evaluation and management. World J Diabetes, 2013, 4: 177-189.
[3] Furie K, Inzucchi SE. Diabetes mellitus, insulin resistance, hyperglycemia, and stroke. Curr Neurol Neurosci Rep, 2008, 8: 12-19.
[4] Adameova A, Dhalla NS, et al. Role of microangiopathy in diabetic cardiomyopathy. Heart Fail Rev, 2014, 19: 25-33.
[5] Hu ZC, Chen YD, Ren YH, et al. Methylprednisolone improves microcirculation in streptozotocin-induced diabetic rats after myocardial ischemia/reperfusion. Chin Med J, 2011, 124: 923-929.
[6] Edvinsson ML, Uddman E, Andersson SE. Deteriorated function of cutaneous microcirculation in chronic congestive heart failure. J Geriatr Cardiol, 2011, 8: 82-87.
[7] Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res, 2010, 106: 1319-1331.
[8] Giacco F, Brownlee M. Oxidative stress and diabetic complications. Cir Res, 2010, 107: 1058-1070.
[9] Mandavia CH, Aroor AR, Demarco VG, et al. Molecular and metabolic mechanisms of cardiac dysfunction in diabetes. Life Sci, 2013, 92: 601-608.
[10] Joshi M, Kotha SR, Malireddy S, et al. Conundrum of pathogenesis of diabetic cardiomyopathy: role of vascular endothelial dysfunction, reactive oxygen species, and mitochondria. Mol Cell Biochem, 2014, 386: 233-249.
[11] Candido R, Forbes JM, Thomas MC, et al. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. Cir Res, 2003, 92: 785-792.
[12] Li X, Luo R, Chen R, et al. Cleavage of IkappaBalpha by calpain induces myocardial NF-kappa B activation, TNF-alpha expression, and cardiac dysfunction in septic mice. Am J Physiol Heart Circ Physiol, 2014, 306: H833-843.
[13] Heidrich FM, Zhang K, Estrada M, et al. Chromogranin B regulates calcium signaling, nuclear factor κB activity, and brain natriuretic peptide production in cardiomyocytes. Cir Res, 2008, 102: 1230-1238. [14] Prasad K. Low Levels of Serum Soluble Receptors for Advanced Glycation End Products, Biomarkers for Disease State: Myth or Reality. Int J Angiol, 2014, 23: 11-16.
[15] Ceradini DJ, Yao D, Grogan RH, et al. Decreasing intracellular superoxide corrects defective ischemia-induced new vessel formation in diabetic mice. J Biol Chem, 2008, 283: 10930-10938.
[16] Thangarajah H, Yao D, Chang EI, et al. The molecular basis for impaired hypoxia-induced VEGF expression in diabetic tissues. Proc Natl Acad Sci USA, 2009, 106: 13505-13510
[17] McQueen AP, Zhang D, Hu P, et al. Contractile dysfunction in hypertrophied hearts with deficient insulin receptor signaling: possible role of reduced capillary density. J Mol Cell Cardiol, 2005, 39: 882-892.
[18] Jesmin S, Zaedi S, Shimojo N, et al.Endothelin antagonism normalizes VEGF signaling and cardiac function in STZ-induced diabetic rat hearts. Am J Physiol Endocrinol Metab, 2007, 292: E1030-1040.
[19] Adameova A, Xu YJ, Duhamel TA, et al. Anti-atherosclerotic molecules targeting oxidative stress and inflammation. Curr Pharm Des, 2009, 15: 3094-3107.
[20] Zhang WF, Xu YY, Xu KP, et al. Inhibitory effect of selaginellin on high glucose-induced apoptosis in differentiated PC12 cells: role of NADPH oxidase and LOX-1. Eur J Pharmacol, 2012, 694: 60-68.
[21] Osto E, Kouroedov A, Mocharla P, et al. Inhibition of protein kinase Cβ prevents foam cell formation by reducing scavenger receptor A expression in human macrophages. Circulation, 2008, 118: 2174-2182.
[22] Wu Y, Wu G, Qi X, et al. Protein kinase C beta inhibitor LY333531 attenuates intercellular adhesion molecule-1 and monocyte chemotactic protein-1 expression in the kidney in diabetic rats. J Pharmacol Sci, 2006, 101: 335-343.
[23] Turner N, Cooney GJ, Kraegen EW, et al. Fatty acid metabolism, energy expenditure and insulin resistance in muscle. J Endocrinol, 2014, 220: T61-79.
[24] Boudina S, Abel ED. Mitochondrial uncoupling: a key contributor to reduced cardiac efficiency in diabetes. Physiology, 2006, 21: 250-258.
[25] Zhang DM, Chai Y, Erickson JR, et al. Intracellular signalling mechanism responsible for modulation of sarcolemmal ATP-sensitive potassium channels by nitric oxide in ventricular cardiomyocytes. J Physiol, 2014, 592: 971-990.
[26] Aljofan M, Ding H. High glucose increases expression of cyclooxygenase-2, increases oxidative stress and decreases the generation of nitric oxide in mouse microvessel endothelial cells. J Cell Physiol, 2010, 222: 669-675.
[27] Rijzewijk LJ, van der Meer RW, Smit JW, et al. Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardio, 2008, 52: 1793-1799.
[28] Kumar S, Prasad S, Sitasawad SL. Multiple antioxidants improve cardiac complications and inhibit cardiac cell death in streptozotocininduced diabetic rats. PLoS One, 2013, 8: e67009.
[29] Adameova A, Abdellatif Y, Dhalla NS. Role of the excessive amounts of circulating catecholamines and glucocorticoids in stress-induced heart disease. Can J Physiol Pharmacol, 2009, 87: 493-514.
[30] 吴婷婷, 魏庆庆, 严宇鹏, 等. 缬沙坦通过下调X-盒结合蛋白1表达抑制糖尿病心肌病大鼠心肌细胞凋亡. 中国循环杂志, 2014, 29: 836-840.
[31] Versari D, Daghini E, Virdis A, et al. Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes Care, 2009, 32: S314-S321.
[32] Jansson PA. Endothelial dysfunction in insulin resistance and type 2 diabetes. J Intern Med, 2007, 262: 173-183.
[33] Kalani M. The importance of endothelin-1 for microvascular dysfunction in diabetes. Vasc Health Risk Manag, 2008, 4: 1061-1068.
[34] Ergul A. Endothelin-1 and diabetic complications: focus on the vasculature. Pharmacol Res, 2011, 63: 477-482.
[35] Arikawa E, Cheung C, Sekirov I, et al. Effects of endothelin receptor blockade on hypervasoreactivity in streptozotocin-diabetic rats: vessel-specific involvement of thromboxane A2. Can J Physiol Pharmacol, 2006, 84: 823-833.
[36] Widyantoro B, Emoto N, Nakayama K, et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation, 2010, 121: 2407-2418.
[37] Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev, 2007, 87: 315-424.
[38] Szabo C. Role of nitrosative stress in the pathogenesis of diabetic vascular dysfunction. Br J Pharmacol, 2009, 156: 713-727.
2014-08-10)
(编辑:许 菁)
266003 山东省青岛市,青岛大学医学院附属医院 老年医学科
颜贵英 硕士研究生 主要从事老年心血管病研究 Email:qingyuefengling@126.com
毛拥军 Email:mmc168@126.com
R54
A
1000-3614(2015)05-0505-03
10.3969/j.issn.1000-3614.2015.05.022
