肥厚型心肌病致病基因型与临床表现的关系及基因筛查在肥厚型心肌病筛查及疾病鉴别诊断中的作用*
2015-12-15崔宏丽王东冯新星邹玉宝王怡璐王继征惠汝太宋雷赵鹏
崔宏丽,王东,冯新星,邹玉宝,王怡璐,王继征,惠汝太,宋雷,赵鹏
肥厚型心肌病致病基因型与临床表现的关系及基因筛查在肥厚型心肌病筛查及疾病鉴别诊断中的作用*
崔宏丽,王东,冯新星,邹玉宝,王怡璐,王继征,惠汝太,宋雷,赵鹏
目的:分析肥厚型心肌病(HCM) 致病基因型与临床表现的关系及基因筛查在HCM筛查及疾病鉴别诊断中的作用。
方法:选择一个HCM家系共14人,多重靶向测序技术对先证者的26个已知最常见的HCM致病基因进行全外显子捕获测序,用Sanger测序对发现的突变进行验证并对其他家系成员及307名健康对照进行该突变位点的筛查,分析其基因型与临床表型的特点。
结果:先证者及其子携带MYH7基因c.2146 G>A(Gly716Arg) 突变,该突变位于MYH7基因19号外显子,导致第716位氨基酸残基由Gly变为Arg,其他25个基因未发现突变。Sanger测序验证后对其他家系成员进行突变筛查,其他家庭成员及对照组未发现该突变,该突变与HCM在该家系中共分离。先证者携带的致病突变为从头突变,并遗传给其子。先证者临床表现为发病早(14岁)、劳力性呼吸困难、胸痛、心悸、心力衰竭,其子出生时即发现心肌肥厚。其父虽然室间隔肥厚(15 mm),但结合其年轻时曾为运动员的经历及遗传筛查的阴性结果,可基本排除其为HCM患者,考虑为生理性肥厚。
结论:该家系HCM由MYH7基因从头突变p.Gly716Arg导致,该突变临床发病早,症状较重,预后较差,为恶性突变。基因筛查在HCM家系筛查及疾病鉴别诊断中有重要意义。
肥厚型心肌病;MYH7基因;Gly716Arg;鉴别诊断
Methods: A HCM pedigree including 14 family members were studied. There were 26 known common HCM related genes were comprehensively screened for mutations in the proband with targeted resequencing. The mutation was identified by bi-directional Sanger sequencing, the specific mutation was examined in all family member and 307 healthy subjects. The relationship between the genotype and the phenotype was studied in this pedigree.
Results: Genetic testing identified a missense mutation, c. 2146 G>A (Gly716Arg) in 19thexon of β-myosin heavy chain gene (MYH7) in proband and her son. No other mutation was found in the rest of 25 genes. No same mutation was found in other family member and 307 healthy subjects. The proband carries a mutation causing HCM, it is a de novo mutation, and the mutation has been transmitted to her son. The proband had the early onset at 14
years of age, suffering from exertional dyspnea, chest pain, palpitation and heart failure; her son expressed HCM at new born. Although proband’s father having ventricular septal hypertrophy at 15mm, taking his athlete career and negative result of genetic screening, he could be basically excluded for HCM and considered as physiological cardiac hypertrophy.
Conclusion: A de novo mutation, Gly716Arg in MYH7 gene caused HCM in a specific Chinese pedigree. This mutation was malignant with early onset, severe symptoms and poor prognosis. Genetic screening is important for differential diagnosis of HCM.
(Chinese Circulation Journal, 2015,30:149.)
肥厚型心肌病(HCM)是最常见的单基因遗传性心血管病,发病率为1/500[1]。该疾病临床表现多样,从无症状到呼吸困难、晕厥、心律失常、心力衰竭不尽相同,最恶劣的并发症为心原性猝死(SCD),发生率约为0.8%[2]。HCM患者由于室间隔异常增厚使得左心室收缩时二尖瓣叶前移与室间隔的贴靠造成左心室流出道狭窄,射血量减少,导致活动时胸闷、晕厥等临床症状,严重者可发生SCD[3],发生机制已证实多是由于室性心动过速或心室颤动所引起[4]。HCM为35岁以下青年人和运动员发生猝死的最主要原因, 给家庭和社会造成了重大损失[5]。大多数HCM患者为编码心肌肌小节蛋白及其相关蛋白的基因突变引起的单基因常染色体显性遗传病[6]。最新的美国心脏病学会基金会/美国心脏协会(ACCF/ AHA)HCM诊治指南推荐对HCM患者进行致病基因检测,遗传检测有助于疾病确诊和家系成员中患者的早期诊断[7]。但HCM有很强的临床和遗传异质性,目前尚缺少足够的基因型—表型关联,导致遗传检测在HCM预后判断中的作用有限[2]。因此,通过家系分析,建立致病突变与临床表型联系,对于揭示HCM的遗传特点、判断预后、指导治疗具有非常重要的意义。在本研究中,我们应用靶向重测序技术在一个HCM家系中对26个已知的最常见HCM致病基因进行了全面筛查,明确了该家系的致病基因和致病突变,分析了该致病突变导致的HCM的临床特点,并且探讨了基因筛查对于HCM家系成员筛查及疾病鉴别诊断的意义。
1 资料与方法
研究对象:2010-01 至2011-10收集的一个HCM家系共14人。其中男性8例,女性6例,年龄4~68岁,平均年龄(39.1±18.6)岁。先证者一位,女性,32岁。HCM诊断标准:成人超声心动图检查室间隔或心室壁厚度≥13 mm,排除长期难以控制的高血压、主动脉瓣狭窄等引起继发性心肌肥厚的心血管系统疾病和全身性疾病。选取307例心电图和心动超声图检查未示异常的健康志愿者作为对照,其中男性194例,女性113例,平均年龄(53.1±10.7)岁。本研究经阜外心血管病医院伦理委员会批准实施。
采集HCM家系的临床资料:体格检查包括身高、体重、血压、心率等;心电图、二维及多普勒超声心动图检查。
基因组DNA提取:采集受调查者外周静脉血4 ml,四乙酸二氨基乙烷(EDTA)抗凝。利用血液基因组提取试剂盒(货号DP319,天根生化科技有限公司,北京) 提取基因组DNA。
先证者基因测序:检测的26个致病基因包括:MYH7, MYBPC3, TNNT2, TNNI3, MYL2, MYL3,TPM1, ACTC1, MYH6, TNNC1, TTN, ACTN2,TCAP, VCL, ANKRD1, CAV3, CSPR3, LDB3,MYOZ2, NEXN, JPH2, PLN, CASQ2, CALR3,PRKAG2 和LAMP2。利用高通量测序法对先证者的26个致病基因的编码外显子和上下游5 bp内含子序列进行靶向测序,基因组DNA经超声破碎为250 bp左右长度片段,利用AMPureXP磁珠(Agencourt,Beckman Coulter, CA, USA)富集200~300 bp范围内的片段。连接通用接头后,聚合酶链式反应(PCR)扩增8个循环,利用定制的Agilent液相捕获文库(Agilent Technologies, Santa Clara, CA, USA)富集目标区域。富集后的基因组片段经过10个循环括增后,Illumina GA IIx (Illumina Inc, CA, USA)进行双端测序,每端读长120 bp。测序数据利用PICARD去除重复后,利用CLC Genomics Workbench (CLC-bio,Aarhus, Denmark)与人类基因组参考序列(GRCh37/ hg19)进行比对,分析26个致病基因目标区域的
覆盖度和变异。变异须满足所在位点测序深度≥25×,并且变异碱基所占比例≥20%。
家系及健康对照的特定基因测序:应用Oligo 6.0软件进行引物设计,由生工生物工程(上海)股份有限公司合成。因MYH7第19号外显子较短,故设计引物时将其与第20号外显子合并在一起。PCR扩增MYH7基因第(19+20)号外显子,引物上游序列:5' CAA AGC CAG GAT CAG AAC CCA G 3';下游序列:5' GGA GTC AAT GGA AAA GAG ATG T 3'。PCR扩增体系20 μl:含基因组DNA 20 ng,上下游引物各0.8 pmol,2×Taq MasterMix(北京康为世纪生物科技有限公司)10 μl。扩增条件:预变性,95℃ 10 min;变性95℃ 30 s,退火52℃30 s,延伸72℃ 40 s,共35个循环;最后延伸72℃,10 min。(PCR仪为FACS Calibur 、美国 BD Biosciences公司产品)
PCR产物纯化后,按双脱氧末端终止法(Sanger法)进行测序( 美国Applied Biosystems公司 3730XL测序仪),测序引物与PCR引物相同。第(19+20)号外显子的目的片段大小约514 bp。
生物信息分析:变异在人群中的频率通过检索Exome Sequencing Project 数据库(ESP,http:// evs.gs.washington.edu/EVS)和1 000 Genomes数据库(http://www.1000genomes.org)确定。变异致病性通过PolyPhen-HCM(www.http://genetics.bwh.harvard.edu/ hcm)预测。
2 结果
HCM家系(图1)的临床及遗传学特点:先证者及其子携带Gly716Arg突变,均呈现临床肥厚型心肌病表现(图2)。①先证者(Ⅲ-6),女性,32岁,14岁时因发热入院,心电图提示ST-T改变。1997年确诊为HCM,主要症状为劳力性呼吸困难、胸痛,当时超声检查示:非对称性肥厚型心肌病(非梗阻型),室间隔中下段明显肥厚(20 mm),左心室舒张末期内径36 mm,左心室射血分数67%,左心房内径30 mm。2007年分娩时出现心力衰竭,之后出现心悸、头晕等症状,无黑矇、晕厥。2014年复查超声:室间隔厚25 mm,左心室舒张末期内径44 mm,左心室射血分数54%,左心房内径40 mm(图2A)。②先证者之子(Ⅳ-3),4岁,出生时心肌即有肥厚(出生时临床资料缺失)。两岁时超声检查示:室间隔明显增厚(15 mm),左心室舒张末期内径29 mm,左心室射血分数78%,左心房内径25 mm,左冠状动脉内径增宽。安静心电图:室内传导阻滞,T波倒置(下壁),ST-T异常(侧壁),异常Q波。动态心电图:窦性心律不齐,窦房结内游走伴不齐,异常Q波,ST-T改变,心率变异性:正常R-R间期标准差>50 ms。2014年复查超声:室间隔厚16 mm,左心室舒张末期内径32 mm,左心室射血分数75%,左心房内径26 mm(图2B)。③先证者之父(Ⅱ-1),无症状,超声检查示:室间隔厚15 mm,左心室舒张末期内径40 mm,左心室射血分数68%,左心房内径25 mm,超声心动图正常(图2C)。基因筛查未发现突变。结合其年轻时曾为运动员的经历,考虑为生理性肥厚,并无临床症状,无需治疗。④ 家系成员中的Ⅱ-3有不典型性胸痛、心悸等临床表现,Ⅱ-5有不典型胸痛的临床表现,但是两者的超声检查均未发现心肌肥厚,心电图检查亦无异常,基因筛查也没有检测到突变。其余家系成员的临床表现和各项检查均无异常。

图1 肥厚型心肌病家系图谱
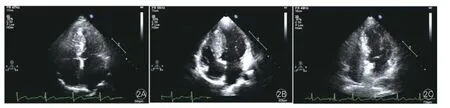
图2 先证者及其父、其子的超声心动图
HCM家系基因测序结果:先证者及其子携带MYH7基因c.2146 G>A(Gly716Arg) 突变(图3A)。该突变位于MYH7基因19号外显子,导致第716位氨基酸残基由Gly变为Arg,其他25个基因未发现突变。Sanger测序验证后对其他家系成员进行突变筛查(图1、3),其他家庭成员及对照组未发现该突变,同源性比对发现MYH7基因Pro716氨基酸残基在不同物种间高度保守(图3B),该突变与HCM在该家系中共分离。
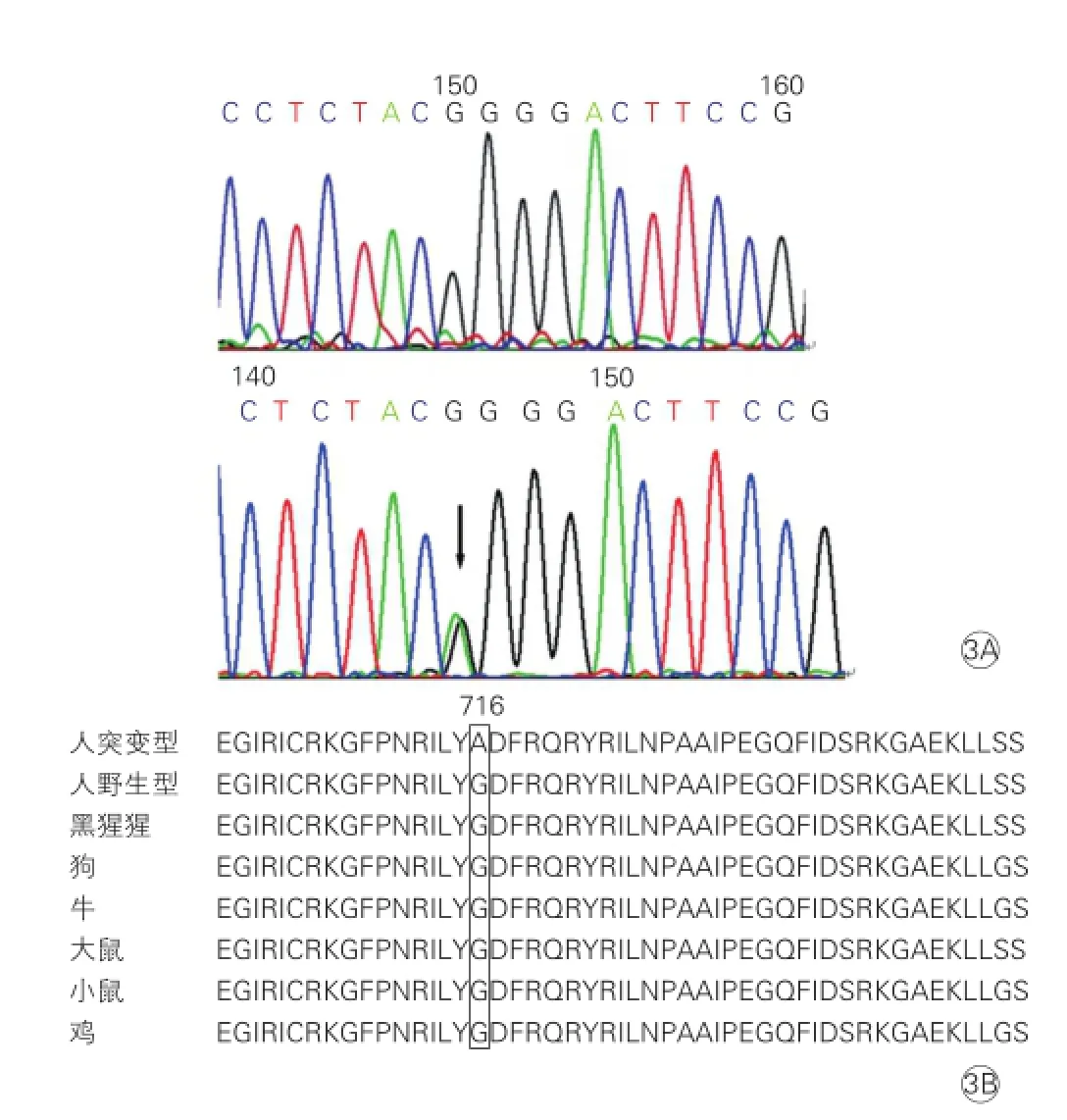
图3 MYH7基因Gly716Arg突变测序图及物种间保守性分析
3 讨论
MYH7的突变所引起的HCM占所有病例的35%~50%,它是第一个被确定的也是最常见的HCM致病基因之一[8]。它编码的蛋白是含1935个氨基酸的肌球蛋白重链-β-MHC。研究发现,发生在MYH7基因上的突变并不是均匀普遍的分布的,也就是说,有聚集性,好发部位主要是β-MHC头部的5个区域,而位于其他部位的突变则没有那么常见[9,10]。
MYH7基因Gly716Arg变异在ESP和1000 Genomes多态数据库中均无报道。同源性比对发现MYH7基因Pro716氨基酸残基在不同物种间高度保守。MYH7基因Gly716Arg突变位于19号外显子,19号外显子编码对应β-MHC头部的靠近铰链区的2个半胱氨酸连接区,发生在此区的突变还包括Arg719Trp、Arg719Gln、Arg723Gly和Gly741Arg。该部位为β-MHC的重要功能区,发生突变可以增加肌球蛋白S1的ATP酶活性,推断其致病机制为阻碍了肌球蛋白的构象改变或改变了肌球蛋白与肌动蛋白及其他分子的相互作用,引起严重的临床表现[11]。
Gly716Arg突变外显率高,携带者发病早、进行性心力衰竭、猝死率高,为恶性突变。Rai等[11]的研究发现此突变携带者具有严重的不对称性室间隔肥厚(31 mm),先证者在21岁时发生猝死。在Anan等[12]做的一项研究中,携带此突变的家系成员发病早,进行性左心室壁变薄、收缩功能减退,顽固性心力衰竭。Arg719Trp、Arg719Gln、Arg723Gly和Gly741Arg突变引起的表型也有相同的恶性临床表现[12-17]。
HCM一直被认为是遵循常染色体显性遗传模式,患者的一级亲属均有50%的可能携带有与患者相同的致病基因,并存在发病的潜在风险[6]。传统的对于HCM的诊断以及猝死危险的评估首先都是基于出现左心室肥厚,但有很大局限性。而基因筛查能够为筛选出家系中有发病可能的亲属提供较为可靠的证据并实现HCM与拟表型疾病(如Fabry病、LEOPARD综合征)的鉴别[18,19]。它还能解决HCM与运动员型心脏以及高血压心肌肥厚的鉴别难题[20,21]。
本研究中先证者之父超声示心肌肥厚(室间隔厚15 mm),符合HCM诊断标准,若不做基因筛查,则极可能依据超声及家族史而诊断为HCM。但先证者及其子携带的致病突变并未在他身上检测到,再结合其年轻时长期从事高强度运动的经历,综合考虑其心肌肥厚为生理性肥厚,现无临床表现故无需进行治疗,仅进行随访观察即可。
目前国内还没有关于Gly716Arg突变的报道,本研究发现的汉族家族性HCM中两例此突变携带者皆发病早(先证者14岁,其子出生即发现心肌肥厚),先证者心肌中度肥厚、劳力性呼吸困难,年纪尚轻就发生心力衰竭,与国外报道的恶性表现基本相符,应对携带者进行积极的观察和治疗,预防猝死的发生。
值得强调的是只有50%的先证者在基因筛查中被检测到,说明仍然有很多与HCM相关的基因未被识别而未能被引入到候选基因范畴[22],并且有时先证者携带有两个致病突变但仅查出一个,或者出现了从头突变,都会影响到基因检测的结果[23]。仍需要大规模多中心临床研究为同时基于基因检测和临床表型的HCM治疗和预防策略的制定提供充分的依据。
本研究中,先证者携带的突变为从头突变,即先证者的父母并不携带突变且无HCM的临床表现。它的发生率是非常低的,徐荣等[24]于2003年报道了中国首例从头突变。从头突变进一步证明了MYH7基因为HCM的责任基因。本研究中先证者之子也遗传了该突变,由此看来,散发性肥厚型心肌病也应警惕其将突变传递给下一代的风险。
这是国内较早发现的MYH7基因Gly716Arg突变,属恶性突变。本研究进一步说明了基因筛查对于HCM家系筛查及疾病鉴别诊断的重要意义以及应警惕散发性肥厚型心肌病将其突变传递给下一代的风险。
[1] Maron BJ. Hypertrophic cardiomyopathy: a systematic review. J Am Meu Assc, 2002, 287: 1308-1320.
[2] Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy related death: revisited in a large non-referral-based patient population. Circulation, 2000, 102: 858-864.
[3] 侯翠红, 乔树宾, 楚建民, 等. 肥厚型梗阻性心肌病行左心室流出道疏通术与经皮室间隔化学消融术治疗的远期疗效分析. 中国循环杂志, 2010, 25: 38-40.
[4] 崔彬, 许建屏, 王巍. 肥厚型梗阻性心肌病围手术期心律失常特点及治疗策略. 中国循环杂志, 2011, 26: 129-132.
[5] Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol, 2012, 60: 705-715.
[6] Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet, 2004, 363: 1881-1891.
[7] Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation, 2011, 124: e783-831.
[8] Tanjore R, RangaRaju A, Vadapalli S, et al. Genetic variations of β-MYH7 in hypertrophic cardiomyopathy and dilated cardiomyopathy. Indian J Hum Genet, 2010, 16: 67-71.
[9] Richard P, Charron P, Carrier L, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for amolecular diagnosis strategy. Circulation, 2003, 107: 2227-2232.
[10] Yu B, Sawyer NA, Caramins M, et al. Denaturing high performance liquid chromatography: high throughput mutation screening in familial hypertrophic cardiomyopathy and SNP genotyping in motor neurone disease. J Clin Pathol, 2005, 58: 479-485.
[11] Rai TS, Ahmad S, Bahl A, et al. Early expression of a malignant phenotype of familial hypertrophic cardiomyopathy associated with a Gly716Arg myosin heavy chain mutation in a Korean family. Am J Cardiol, 1998, 82: 1509-1513.
[12] Anan R, Greve G, Thierfelder L, et al. Prognostic implications of novel beta cardiac myosin heavy chain gene mutations that cause familial hypertrophic cardiomyopathy. J Clin Invest, 1994, 93: 280-285.
[13] 宋雷, 黄晓红, 惠汝太, 等. 心肌球蛋白重链基因Arg719Gln突变与家族性肥厚型心肌病. 中华心血管病杂志, 2001, 29: 348-352.
[14] Enjuto M, Francino A, Navarro-López F, et al. Malignant hypertrophic cardiomyopathy caused by the Arg723Gly mutation in beta-myosin heavy chain gene. J Mol Cell Cardiol, 2000, 32: 2307-2313.
[15] 郑冬冬, 杨俊华, 董宁征, 等. 中国汉族家族性肥厚型心肌病人群MYH7基因Arg723Gly突变分析. 中华心血管病杂志, 2006, 34: 208-211.
[16] Dӧhlemann C, Hebe J, Meitinger T, et al. Apical hypertrophic cardiomyopathy due to a de novo mutation Arg719Trp of the betamyosin heavy chain gene and cardiac arrest in childhood. A case report and family study. Z Kardiol, 2000, 89: 612-619.
[17] Miller G, Colegrave M, Peckham M. N232S, G741R and D778G beta-cardiac myosin mutants, implicated in familial hypertrophic cardiomyopathy, do not disrupt myofibrillar organisation in cultured myotubes. FEBS Lett, 2000, 486: 325-327.
[18] O'Mahony C, Lambiase PD, Quarta G, et al. The long-term survival and the risks and benefits of implantable cardioverter defibrillators in patients with hypertrophyic cardiomyopathy. Heart, 2012, 98: 116-125.
[19] Weidemann F, Niemann M, Breunig F, et al. Long-term effects of enzyme replacement therapy on Fabry cardiomyopathy Evidence for a better outcome with early treatment. Circulation, 2009, 119: 524-529.
[20] Patel MR, Cecchi F, Cizmarik M, et al. Cardiovascular events in patients with Fabry disease natural history data from the Fabry registry. J Am Coll Cardiol, 2011, 9: 1093-1099.
[21] Andersen PS, Havndrup O, Hougs L, et al. Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum Mutat, 2009, 30: 363-370.
[22] Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Heart Rhythm, 2011, 8: 1308-1339.
[23] Erdmann J, Daehmlow S, Wischke S, et al. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin Genet, 2003, 64 : 339-349.
[24] 徐荣, 王继征, 邹玉宝, 等. 心肌肌钙蛋白I基因Arg170Gln突变—de novo突变与肥厚型心肌病. 中华医学杂志, 2003, 83: 1634.
Relationship Between the Genotype Causing Hypertrophic Cardiomyopathy With its Clinical Presentation and Differential Diagnosis
CUI Hong-li, WANG Dong, FENG Xin-xing, ZOU Yu-bao, WANG Yi-lu, WANG Ji-zheng, HUI Ru-tai, SONG Lei, ZHAO Peng. Department of Pathology, Affliated Hospital, Qingdao University Medical College, Qingdao (266003), Shandong, China
Co-corresponding Authors: SONG Lei, Email: lsongqd@yahoo.com and ZHAO Peng, Email: zhpeng17@hotmail.com
Objective: To identify the mutation gene for Chinese Han population with hypertrophic cardiomyopathy (HCM), to analyze the relationship between the genotype and phenotype, and to study the effect of genetic screening in HCM pedigrees for differential diagnosis of HCM.
Hypertrophic cardiomyopathy; MYH7gene; Gly716Arg; Differential diagnosis
2014-03-12)
(编辑: 曹洪红)
青岛市产学研合作引导计划(应用基础研究)(13-1-4-141-jch)
266003 山东省青岛市,青岛大学医学院附属医院 病理科(崔宏丽、赵鹏);中国医学科学院 北京协和医学院 国家心血管病中心阜外心血管病医院 心血管疾病国家重点实验室(王东、冯新星、邹玉宝、王怡璐、王继征、惠汝太、宋雷)
崔宏丽 硕士研究生 研究方向为肥厚型心肌病与心原性猝死 Email:cui1998322@163.com 通讯作者:宋雷 Email:lsongqd@yahoo.com赵鹏 Email:zhpeng17@hotmail.com
R541
A
1000-3614(2015)02-0149-05
10.3969/j.issn.1000-3614.2015. 02.014
