页岩有机质孔缝内液态烷烃赋存状态分子动力学模拟
2015-12-07王森冯其红查明卢双舫秦勇夏天张驰
王森,冯其红,查明,卢双舫,秦勇,夏天,张驰
(1.中国石油大学(华东);2.The University of Texas at Austin;3.中国石油勘探开发研究院;4.中国石油大学(北京))
页岩有机质孔缝内液态烷烃赋存状态分子动力学模拟
王森1,2,冯其红1,查明1,卢双舫1,秦勇3,夏天1,张驰4
(1.中国石油大学(华东);2.The University of Texas at Austin;3.中国石油勘探开发研究院;4.中国石油大学(北京))
利用分子动力学模拟方法研究页岩有机质孔缝内液态烷烃赋存状态。基于OPLS(Optimized Potentials for Liquid Simulation)力场模拟计算了烷烃在不同温度和压力条件下的密度,并与实验值比较,验证了该方法的有效性。在此基础上,以正庚烷为例分析了油藏条件下有机质孔缝内烷烃赋存的基本特征,并探讨了缝宽、有机质成熟度、烷烃碳链长度以及同分异构体对其赋存的影响。结果表明:①烷烃在有机质孔缝内的密度分布并非均匀,而是呈现周期性的波动;②在靠近固体壁面处烷烃会形成“类固体”层,其密度约为游离态流体密度的1.9~2.7倍;③液态烷烃在有机质孔缝内主要发生多层吸附,每个吸附层的厚度约为0.48 nm,吸附层的数目受缝宽和流体组分影响。实例模拟分析表明,原油在有机质孔缝内以吸附态形式存在的比例为18.2%。图9表2参24
页岩油;有机质;液态烷烃;赋存状态;烷烃密度;吸附层
0 引言
页岩油的储集空间类型主要包括有机质孔缝、基质晶间孔、粒间孔和粒内孔等[1-5]。其中有机质孔缝是页岩储集空间的特色和重要组成部分。与其他矿物相比,有机质与液态烷烃之间具有较强的相互作用,因此有机质对页岩油的赋存状态具有较大影响[2,6]。本文以页岩油为研究对象,利用分子动力学模拟方法开展了有机质孔缝内烷烃赋存状态的研究,分析了孔缝尺寸、有机质热成熟度、烷烃碳链长度以及同分异构体等因素对原油赋存状态的影响,以期为页岩内原油的可动性评价提供新的思路和方法。由于在相同尺寸下狭缝与孔缝内流体的赋存状态差别不大[7],因此为了提高模拟效率,用狭缝代替孔缝对页岩油的赋存状态进行研究。这也是目前国际学术界常用的处理方法[8-9]。
1 烷烃赋存状态模拟的力场模型
分子动力学利用牛顿运动定律模拟多体系统内原
子或分子的运动轨迹,并通过对其不同状态所构成的系综(Ensemble)进行统计平均计算体系的结构和性质。该方法的准确性取决于力场模型的筛选,但不同物质所适用的力场不尽相同。对于烷烃,目前国际上一般采用OPLS(Optimized Potentials for Liquid Simulation)力场进行描述[8,10]。该力场的突出特点在于不同原子之间的势能参数是通过对室内实验结果拟合得到的,因此可靠性高。该力场包括全原子力场(OPLS-AA)和粗粒化力场(OPLS-UA)两种类型(见图1),其差别在于利用OPLS-AA力场进行模拟时,分子内各原子的力场参数都需要提供,每个原子都参与计算并在其他原子的作用下发生运动;但在OPLS-UA力场中,烷烃分子上的—CH3和—CH2—基团分别被视为单个的原子并被整体赋予相应的力场参数,因此模拟计算量将大大减少。
基于OPLS-AA和OPLS-UA力场,本文分别模拟计算了正庚烷在不同温度(70~100 ℃)和压力(18~30 MPa)条件下的密度并将模拟结果与实验值对比,从模拟精度和所需时间两方面对力场模型进行比较,筛选出的模型将被用于烷烃在有机质孔缝内赋存状态的模拟。在计算其密度的过程中,首先构建正庚烷的全原子模型和粗粒化模型(见图1),并应用能量最小化方法分别对其结构进行优化得到稳定的分子构型。随后将一定数量的(本模型中为150个)经过构型优化的正庚烷分子放入正方体的模拟盒内,并对体系的能量进行监测,避免发生原子相互重叠或距离太近的情形。然后设置模拟盒在3个方向上均为周期性边界,对整个体系进行能量最小化,随后以1 fs为时间步长,在固定分子数目及定温定压条件下(NPT系综)模拟1 000 ps,其中前500 ps通过调节模拟盒的大小使体系逐渐达到平衡,后500 ps的模拟盒体积用于输出。对输出结果进行算术平均可计算得到实验温度和压力条件下正庚烷的密度[8]。不同条件下的模拟结果及所用时间见表1,其中实验值为美国国家标准技术研究院公布的结果[11]。模拟过程中对两种力场采用同样的设置:范德华力的截断半径为1.2 nm,不同原子之间的非键结势能采用Lorentz-Berthelot混合准则计算。同时为提高计算效率,对OPLS-AA力场采用PPPM(particle–particle particle–mesh)算法计算长程静电力;对OPLS-UA力场,由于—CH3和—CH2—基团不带电,因此不需要计算静电力。模拟在工作站(CPU型号:i7-4770K)上利用美国Sandia国家实验室的大规模原子/分子并行模拟器LAMMPS[12]通过8核并行计算完成。
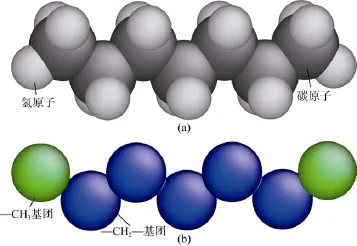
图1 正庚烷的全原子力场(a)和粗粒化力场(b)模型图

表1 不同力场模型下正庚烷密度计算结果与实验值的对比
由表1可知,全原子模型和粗粒化模型均可较为准确地模拟烷烃性质,其中全原子模型的计算结果更接近实验值(相对误差仅为0.169 4%),而粗粒化模型的相对误差稍大(平均5.174 7%)。但是同时可以发现采用粗粒化模型后,模拟相同时间步所需的计算机时间仅为全原子模型的1/9。考虑到对烷烃赋存状态进行研究时,所需的分子数目及时间步数都要大大增加,因此采用粗粒化模型进行烷烃的赋存状态模拟,但本文的方法同样适用于全原子模型。
2 烷烃赋存状态的分子动力学模拟
2.1 模型的建立
页岩中有机质的化学组成非常复杂,如何准确地在分子尺度上对其结构进行解析尚属世界难题。为简
便起见,目前国际上一般采用石墨烯近似代替有机质结构[8-9,13]。为保证固体壁面的厚度大于OPLS力场的截断半径,采用6层石墨烯作为有机质纳米缝的固体壁面。设定每层石墨烯x方向和y方向的尺寸分别为2.952 0 nm和2.556 5 nm,且相邻两层之间相互平行(间距为0.335 nm)。流体结构模型的建立与上文正庚烷密度模拟时的建模过程基本一致,但需采用长方体的模拟盒,且盒子的底面尺寸与固体壁面的表面尺寸保持一致,盒子高度为有机质孔缝的宽度。最后将流体的分子结构模型嵌入纳米孔缝的结构模型中即完成了有机质孔缝内烷烃赋存模拟所需的分子结构模型。
2.2 模拟过程
首先利用共轭梯度算法对体系能量最小化,通过不断调节原子的位置获得稳定的初始构型;然后设置模拟的时间步长为1 fs,采用Nosé–Hoover[8]算法控制体系的温度为油藏温度,在固定分子数目及定体积定温度条件下(NVT系综)模拟1 000 ps使体系达到平衡,当体系的总能量、温度和压力等不随时间变化时认为体系已达到平衡状态;最后在固定分子数目及定体积定能量条件下(NVE系综)模拟2 000 ps,并以1 ps为时间间隔收集数据用于统计分析。模拟过程中构成壁面的碳原子固定不动。
2.3 模拟结果后处理
分子动力学模拟直接得到的是各个原子的运动轨迹,因此必须进一步利用统计热力学方法将模拟结果转换为宏观物理量。为计算有机质孔缝内烷烃的密度分布,首先在与固体壁面平行的方向上将纳米孔缝划分为Ns个单元(本文中每个单元的厚度为0.04 nm),并定义如下标志函数:

则对于第n个单元,从时间步Js到Je的流体密度平均值为:
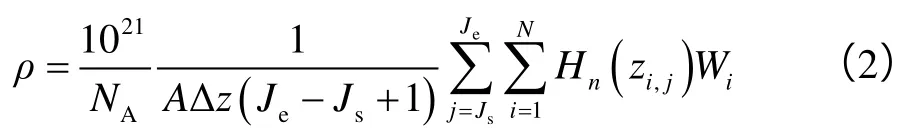
本文的可视化结果采用美国麻省理工学院的公开软件Atomeye实现[14]。
3 有机质孔缝内烷烃赋存的基本特征
图2为80 ℃、30 MPa下正庚烷在宽度为7.8 nm的有机质孔缝内达到平衡后其截面上的密度分布。由图2可见,其密度分布并不均匀,而是出现了周期性的波动。从固体壁面到孔道中央,流体密度的峰值逐渐减小;在靠近孔道中央的位置,由于壁面与流体的相互作用力较小,流体的密度不再变化。将-2~2 nm的流体密度进行算术平均,可得到孔道中央流体的密度为0.671 g/cm3,该值与正庚烷在该条件下的密度实验值(0.666 g/cm3)[11]吻合较好,验证了模拟方法的准确性。由于密度分布曲线上的每个峰值对应于流体在该孔缝内形成的吸附层[8,15],因此正庚烷在距离孔道中央3.63 nm,3.11 nm和2.67 nm的位置处形成了3个对称的吸附层。其中距离壁面最近的吸附层(第1吸附层)密度峰值达到了1.56 g/cm3,约为孔道中央流体密度的2.3倍,因此可以认为该吸附层内的烷烃分子以固体或“类固体”形式存在[8]。该现象在页岩气藏中已经被发现[8],但是对于富液态烃类页岩尚属首次。
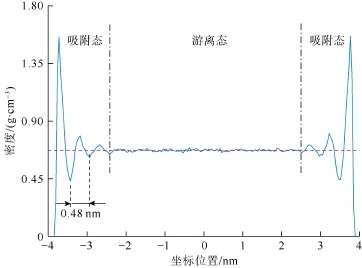
图2 正庚烷在7.8 nm的有机质孔缝内达到平衡后的密度分布
正庚烷在有机质孔缝内赋存的微观结构也进一步证实了该结论。由图3可以看出,在靠近固体壁面处,烷烃分子形成了一个非常明显的吸附层(第1吸附层),而且该吸附层与其他的烷烃分子之间相互分离,该分离位置即对应于图2中密度的极小值。逐渐远离固体壁面,有机质对烷烃分子的吸引力逐渐减小,烷烃分子之间的相互作用力逐渐处于主导位置,因此另两个吸附层的峰值密度较小,在微观结构图上不能观察到明显的分层结构。在孔道中央位置处,烷烃分子呈现无序的随机分布(见图3体相流体)。进一步可以根据密度分布曲线上相邻两个波谷之间的水平距离确定每个吸附层的厚度。对于正庚烷来说,3个吸附层的厚度均为0.48 nm,该数值与正构烷烃分子的宽度一致[16]。由此可以估计出在该有机质孔缝内,占总孔隙体积36.9%(0.48´6/7.8=36.9%)的烷烃吸附于有机质表面,其余的烷烃以游离态形式存在于孔道中央。该结论初
步为邹才能等提出的页岩油滞留聚集模式提供了的支撑[2]。此外,由于孔缝内流体密度分布不均匀,因此可以根据图2求出各吸附层的平均密度进而估算出吸附态烷烃所占的质量分数。因为吸附态流体的密度大于游离态的密度,因此吸附态所占的质量分数大于其体积分数(36.9%)。

图3 正庚烷在宽度为7.8 nm的有机质孔缝内的赋存状态
4 敏感性分析
4.1 孔缝宽度对烷烃赋存状态的影响
图4为不同宽度的有机质孔缝内正庚烷的密度分布。当缝宽为1.62 nm时,孔缝内所有的烷烃分子均吸附于有机质表面且形成了2个对称的吸附层,孔道内不存在游离态的烷烃。当缝宽为3.52 nm时,孔道内形成了3个吸附层,仅在孔道中央存在少量的游离态原油。随缝宽继续增大,吸附层的数目保持不变,游离态原油的含量将逐渐增大。因此,当缝宽小于2.88 nm(0.48´3´2=2.88 nm。每层厚度为0.48 nm,一共3个对称的吸附层)时,缝内的正庚烷全部以吸附态形式存在;当其宽度大于2.88 nm时,烷烃分子在孔缝内形成对称的3个吸附层,每层的宽度为0.48 nm,吸附层以外的流体以游离态形式存在,且游离态流体的密度与相应条件下的实验值基本一致。此外,由图4可知,随着缝宽的增大,吸附层流体的密度峰值也逐渐增大,但是其增大的趋势逐渐变缓,该结论与Chen等人的研究结果一致[17]。当缝宽由1.62 nm增大至11.32 nm时,第1吸附层的密度峰值由1.265 g/cm3增大到1.686 g/cm3,分别为游离态流体密度的1.9倍和2.5倍。继续增大缝宽,正庚烷在有机质孔缝内的吸附层数将稳定在3层,第1吸附层的密度峰值将逐渐增大并最终稳定为游离态流体密度的2.7倍。
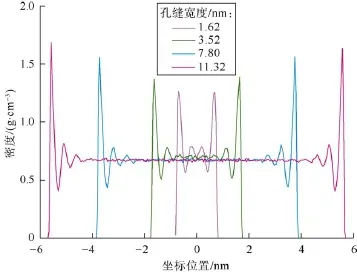
图4 正庚烷在不同宽度有机质孔缝内的密度分布
4.2 有机质热成熟度对烷烃赋存状态的影响
鉴于干酪根在演化过程中化学成分逐渐发生变化,在此初步探讨了有机质热成熟度对烷烃赋存状态的影响。随着热成熟度的升高,有机质中的脂肪链、羧基、羟基和羰基等逐渐消失,氢碳比和氧碳比逐渐减小。Bagri等[18]利用分子动力学模拟方法和第一性原理对比了石墨烯表面添加不同化学基团的稳定性,发现与羟基和环氧基等基团相比,有机质表面更易于形成羰基。因此,笔者通过在石墨烯表面添加不同数目的羰基,近似代替不同成熟度的有机质。羰基的数目越多,氧碳比越高,有机质的成熟度越低。Liu和Wilcox以及Hu等采用类似的处理方式分别研究了有机质表面CO2的吸附及其润湿性的变化[19-20]。
图5为石墨烯表面有羰基存在时的分子结构模型,其中碳氧键的键长为0.123 nm,碳氧键与石墨烯平面之间的夹角为49.5°[19]。通过在石墨烯表面添加不同数目的羰基使有机质模型的氧碳比分别为:0.04,0.08和0.16,该范围与巴黎盆地托森页岩干酪根的热演化过程一致(0.026~0.196)[21]。由图6a可知,不同成熟度的有机质孔缝内正庚烷的密度分布差异不大,仅第1吸附层的峰值密度存在细微差异,因此该条件下
有机质成熟度对烷烃的赋存状态没有明显影响。该结论与Zhang Tongwei等对不同成熟度页岩表面甲烷吸附能力的测试结果基本一致[22]。进一步细致比较可以发现,有机质成熟度较低(氧碳比较高)时,第1吸附层的峰值密度较大(见图6b),表明有机质对烷烃的吸附能力较强,孔隙中以游离态形式存在的烷烃较少。其原因在于,此时干酪根内存在较多的羰基,由于烷烃为非极性分子,有机质与烷烃之间主要靠范德华力起作用,较多的羰基使有机质与烷烃之间具有较强的相互作用力,因此吸附态原油比例较高。随着有机质热成熟度的升高,羰基发生热解反应,数目逐渐减少,因此对烷烃的吸附能力减弱,游离态烷烃的比例增多。当干酪根逐渐转化为石墨时,其吸附能力将稍微增强。该结论或可以解释为何不同研究者得到的关于页岩成熟度对其吸附能力影响的结果截然相反[22-23]。
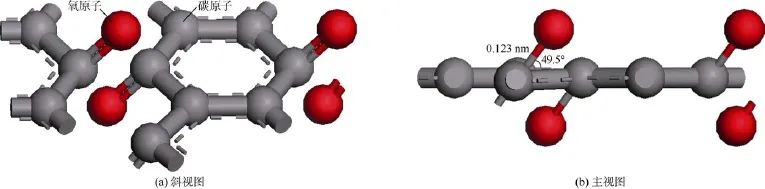
图5 石墨烯表面有羰基存在时的分子结构模型
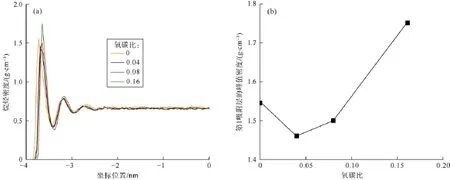
图6 不同成熟度有机质孔缝内烷烃的密度分布(a)及第1吸附层的峰值密度(b)
4.3 烷烃碳链长度对其赋存状态的影响
正庚烷、正癸烷和正十五烷在7.8 nm有机质孔缝内的赋存状态见图7。由图7可见,不同碳链长度的正构烷烃都会在靠近固体壁面处形成一定数量的吸附层,但吸附层的数量和密度峰值与碳链长度有关。碳链越长,烷烃与有机质的相互作用力越强,烷烃分子越容易发生吸附,从而使得吸附层的密度增大,层数增多。由此可以推测,对于富液态烃类页岩,原油中所含有的重质组分越多,其在有机质表面的吸附作用越明显,吸附态原油所占的比例越高,在评价其可动性时越有必要考虑。

图7 不同碳原子数的正构烷烃在有机质孔缝内的密度分布
4.4 烷烃同分异构体对其赋存状态的影响
同分异构现象广泛存在于烷烃中,笔者简要分析
了庚烷的3种同分异构体在有机质孔缝内赋存状态的差异。由图8可知,2-甲基己烷、2,4-二甲基戊烷和3-乙基戊烷这3种同分异构体的密度分布曲线整体特征基本相同,说明支链对烷烃在有机质孔缝内的赋存状态没有明显影响。该结论与Enevoldsen等利用中子散射实验得到的结论一致[24]。此外可以发现该密度分布曲线上较大的差异出现在第2吸附层,即同分异构体的支链数目越多,支链越长,则第2吸附层密度峰值出现的位置距固体壁面越远,该吸附层的厚度也就越大。出现该现象的原因在于当烷烃支链的数目和长度增大时,其结构不再是原来的链状,因此不同分子之间呈现规则排列的难度较大。然而由于第1吸附层与固体壁面之间的相互作用较强,因此同分异构体结构的差异对第1吸附层的影响不明显。
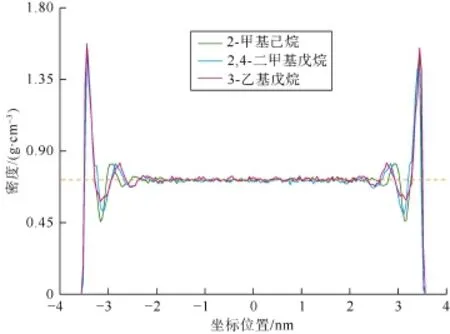
图8 庚烷的3种同分异构体在有机质孔缝内的密度分布
5 实例应用
以上分析中流体为单一组分,下面采用本文方法计算有机质孔缝内烷烃混合物的密度分布,并对有机质孔缝内以吸附态形式存在的原油比例进行计算。原油组分数据见表2,其他参数:油藏温度116 ℃,压力47.2 MPa,孔喉直径15.82 nm。图9为该有机质孔缝内原油的密度分布。由于孔道中央的原油密度基本不变,为体现靠近固体壁面处原油密度的波动,位于-6~6 nm的密度数值没有绘制出来。由图9可知,原油在有机质表面形成了3个对称的吸附层,每个吸附层的厚度同样为0.48 nm,第1吸附层的峰值密度约为1.69 g/cm3,为游离态原油密度(0.611 g/cm3)的2.77倍。由此可估算出该有机质孔缝内吸附态原油占总孔隙体积的比例为18.2%。

表2 原油组分数据

图9 有机质孔缝内原油的密度分布
6 结论
以页岩油为研究对象,基于粗粒化的OPLS力场建立了有机质孔缝内烷烃赋存状态的分子动力学模拟方法。首先利用该方法对不同温度、压力条件下烷烃的密度进行了模拟,计算结果与实验值较为吻合。与全原子力场模型相比,计算效率提高了约9倍,充分验证了该方法的有效性。
利用该方法对页岩油藏有机质孔缝内烷烃赋存状态的基本特征进行了研究。结果表明:烷烃在有机质孔缝内的密度分布并不均匀,而是呈现周期性的波动。在靠近固体壁面处烷烃会形成“类固体”层,其密度为游离态流体密度的1.9~2.7倍;而孔道中央为游离态原油,其密度与相应条件下体相原油密度的实验值一致。液态烷烃在有机质孔缝内主要为多层吸附,每个吸附层的厚度约为0.48 nm,但吸附层的数目受缝宽和流体组分影响。
进一步探讨了孔缝宽度、有机质热成熟度、烷烃碳链长度以及同分异构体对烷烃赋存状态的影响。随着缝宽的增大,吸附层的数目逐渐增多,但当其宽度大于一定值后,吸附层的数目保持不变。随着成熟度的升高,有机质对烷烃的吸附能力减弱。烷烃碳链长度越长,吸附层数越多,吸附相流体密度越大。同分异构体对烷烃的赋存没有明显影响。最后以烷烃混合
物为例进行了计算,结果表明在该有机质孔缝内18.2%(体积分数)的原油以吸附态形式存在。
分子动力学模拟在地质学中的应用刚刚起步,矿物的结构模型和大规模的求解方法尚不成熟。本研究还有很多因素未考虑,如有机质内氮、硫、氢等元素以及其他官能团的影响、多孔介质结构的复杂性、流固耦合的相互作用以及流体相态的变化等,这将会在后续研究中逐步完善。
符号注释:
A——固体壁面表面积,nm2;Hn——标志函数;Js,Je——计算的开始、结束时间步;n——单元的序号,n=1,2,…,Ns;N——模拟体系中构成流体的所有原子的个数;NA——阿伏伽德罗常数,6.022×1023mol-1;Ns——对模拟结果进行后处理时所划分孔缝单元的数目;Wi——原子i的摩尔质量,g/mol;z——原子在z方向的坐标;Δz——孔缝单元的厚度,nm;ρ——密度,g/cm3。下标:i——原子的序号;j——时间步序号。
[1] 张金川,林腊梅,李玉喜,等.页岩油分类与评价[J].地学前缘,2012,19(5): 322-331.Zhang Jinchuan,Lin Lamei,Li Yuxi,et al.Classification and evaluation of shale oil[J].Earth Science Frontiers,2012,19(5): 322-331.
[2] 邹才能,杨智,张国生,等.常规-非常规油气“有序聚集”理论认识及实践意义[J].石油勘探与开发,2014,41(1): 14-27.Zou Caineng,Yang Zhi,Zhang Guosheng,et al.Conventional and unconventional petroleum “orderly accumulation”: Concept and practical significance[J].Petroleum Exploration and Development,2014,41(1): 14-27.
[3] 张林晔,包友书,李钜源,等.湖相页岩油可动性: 以渤海湾盆地济阳坳陷东营凹陷为例[J].石油勘探与开发,2014,41(6): 641-649.Zhang Linye,Bao Youshu,Li Juyuan,et al.Movability of lacustrine shale oil: A case study of Dongying Sag,Jiyang Depression,Bohai Bay Basin[J].Petroleum Exploration and Development,2014,41(6): 641-649.
[4] 蒲泊伶,董大忠,吴松涛,等.川南地区下古生界海相页岩微观储集空间类型[J].中国石油大学学报: 自然科学版,2014,38(4): 19-25.Pu Boling,Dong Dazhong,Wu Songtao,et al.Microscopic space types of Lower Paleozoic marine shale in southern Sichuan Basin[J].Journal of China University of Petroleum: Edition of Natural Science,2014,38(4): 19-25.
[5] 郭小波,黄志龙,陈旋,等.马朗凹陷芦草沟组泥页岩储层含油性特征与评价[J].沉积学报,2014,32(1): 166-173.Guo Xiaobo,Huang Zhilong,Chen Xuan,et al.The oil-bearing property characteristics and evaluation of Lucaogou formation shale reservoirs in Malang Sag[J].Acta Sedimentologica Sinica,2014,32(1): 166-173.
[6] 贾承造,郑民,张永峰.非常规油气地质学重要理论问题[J].石油学报,2014,35(1): 1-10.Jia Chengzao,Zheng Min,Zhang Yongfeng.Four important theoretical issues of unconventional petroleum geology[J].Acta Petrolei Sinica,2014,35(1): 1-10.
[7] Song X,Chen J K.A comparative study on Poiseuille flow of simple fluids through cylindrical and slit-like nanochannels[J].International Journal of Heat and Mass Transfer,2008,51(7): 1770-1779.
[8] Ambrose R J,HartmanR C,Diaz-Campos M,et al.Shale gas-in-place calculations Part I: New pore-scale considerations[J].SPE Journal,2012,17(1): 219-229.
[9] Mosher K,He J J,Liu Y Y,et al.Molecular simulation of methane adsorption in micro-and mesoporous carbons with applications to coal and gas shale systems[J].International Journal of Coal Geology,2013,109: 36-44.
[10] Jorgensen W L,Madura J D,Swenson C J.Optimized intermolecular potential functions for liquid hydrocarbons[J].Journal of the American Chemical Society,1984,106(22): 6638-6646.
[11] National Institute of Standards and Technology.Thermophysical properties of fluid systems[EB/OL].[2014-10-15].http://webbook.nist.gov/chemistry/fluid/.
[12] Plimpton S.Fast parallel algorithms for short-range molecular dynamics[J].Journal of Computational Physics,1995,117(1): 1-19.
[13] Harrison A,Cracknell R F,Krueger-Venus J,et al.Branched versus linear alkane adsorption in carbonaceous slit pores[J].Adsorption,2014,20: 427-437.
[14] Li J.AtomEye: An efficient atomistic configuration viewer[J].Modelling and Simulation in Materials Science and Engineering,2003,11(2): 173-177.
[15] Sha M,Zhang F,Wu G,et al.Ordering layers of [bmim][PF6] ionic liquid on graphite surfaces: Molecular dynamics simulation[J].The Journal of Chemical Physics,2008,128(13): 134504-1-134504-7.
[16] Christenson H K,Gruen D W R,Horn R G,et al.Structuring in liquid alkanes between solid surfaces: Force measurements and mean-field theory[J].The Journal of Chemical Physics,1987,87(3): 1834-1841.
[17] Chen X Y,Liu Y,Yang J M.Density oscillation from nanoscale to macroscale[J].Modern Physics Letters B,2008,22: 2649-2658.
[18] Bagri A,Grantab R,Medhekar N V,et al.Stability and formation mechanisms of carbonyl-and hydroxyl-decorated holes in grapheneoxide[J].The Journal of Physical Chemistry C,2010,114(28): 12053-12061.
[19] Liu Yangyang,Wilcox J.Effects of surface heterogeneity on the adsorption of CO2in microporous carbons[J].Environmental Science &Technology,2012,46(3): 1940-1947.
[20] Hu Y,Devegowda D,Sigal R F.Impact of maturity on kerogen pore wettability: A modeling study[R].SPE 170915,2014.
[21] Behar F,Vandenbroucke M.Chemical modelling of kerogens[J].Organic Geochemistry,1987,11(1): 15-24.
[22] Zhang Tongwei,Ellis G S,Ruppel S C,et al.Effect of organic-matter type and thermal maturity on methane adsorption in shale-gas systems[J].Organic Geochemistry,2012,47: 120-131.
[23] 张寒,朱炎铭,夏筱红,等.页岩中有机质与黏土矿物对甲烷吸附能力的探讨[J].煤炭学报,2013,38(5): 812-816.Zhang Han,Zhu Yanming,Xia Xiaohong,et al.Comparison and explanation of the absorptivity of organic matters and clay minerals in shales[J].Journal of China Coal Society,2013,38(5): 812-816.
[24] Enevoldsen A D,Hansen F Y,Diama A,et al.Comparative study of normal and branched alkane monolayer films adsorbed on a solid surface II: Dynamics[J].The Journal of Chemical Physics,2007,126(10): 104704-1-104704-10.
(编辑 郭海莉)
Molecular dynamics simulation of liquid alkane occurrence state in pores and fractures of shale organic matter
Wang Sen1,2,Feng Qihong1,Zha Ming1,Lu Shuangfang1,Qin Yong3,Xia Tian1,Zhang Chi4
(1.China University of Petroleum (East China),Qingdao 266580,China;2.The University of Texas at Austin,Austin 78713,United States;3.PetroChina Research Institute of Petroleum Exploration &Development,Beijing 100083,China;4.China University of Petroleum,Beijing 102249,China)
Molecular dynamics simulation was used to study the occurrence state of liquid alkane in pores and fractures of shale organic matter.On the basis of OPLS (Optimized Potentials for Liquid Simulation) force field,the alkane densities under different pressures and temperatures were calculated;the comparison with experimental results validated the efficiency of this approach.With n-heptane as an example,the basic occurrence behaviors of alkanes in the pores and fractures of organic matter were analyzed under formation conditions,and the effects of fracture width,thermal maturity of organic matter,carbon chain length and isomers were also discussed.Results show that: (1) The density distribution of the alkanes across the pores and fractures is not uniform,but presents a periodic fluctuation;(2) A“solid-like” alkane layer will form in the vicinity of the solid surface,and its density approximates to 1.9-2.7 times greater than that of the bulk-fluid;(3) Multiple adsorption layers are always shown for liquid alkanes and the thickness of each layer is 0.48 nm;the total number of adsorbed layers is influenced by the fracture width and fluid composition.Finally,using this approach,the proportion of adsorbed-phase (18.2%) is determined for oil in an organic matter slit.
shale oil;organic matter;liquid hydrocarbon;occurrence state;alkane density;adsorption layer
国家重点基础研究发展计划“中国陆相致密油(页岩油)形成机理与富集规律”(2014CB239005);国家自然科学基金重点基金(41330313);教育部长江学者和创新团队发展计划(IRT1294);中国石油大学(华东)优秀博士学位论文培育计划(LW140201A);中国石油大学(华东)研究生创新工程(YCX2014009)
TE122
A
1000-0747(2015)06-0772-07
10.11698/PED.2015.06.10
王森(1986-),男,河南南阳人,中国石油大学(华东)在读博士,现为美国德克萨斯大学Austin分校联合培养博士研究生,主要从事非常规油气勘探开发研究。地址:山东省青岛市黄岛区长江西路66号,中国石油大学(华东)石油工程学院,邮政编码:266580。E-mail: wangsena1@126.com
联系作者:冯其红(1969-),男,四川西充人,博士,中国石油大学(华东)教授、博士生导师,主要从事非常规油气勘探开发及提高采收率方面的科研工作。地址:山东省青岛市黄岛区长江西路66号,中国石油大学(华东)石油工程学院,邮政编码:266580。E-mail: fengqihong@126.com
2015-01-12
2015-10-12
