肝豆状核变性合并肝脏损害的临床误诊分析
2015-12-06袁兴娅甘华田
袁兴娅,熊 海,甘华田
1.四川大学华西第四医院(成都 610041);2.四川大学华西医院(成都 610041)
肝豆状核变性(hepatolenticular degeneration,HLD),是一种以原发性铜代谢障碍为特征的常染色体隐性遗传病,其中肝脏是HLD铜沉积和损害的主要靶器官之一,最常见的肝脏损害首发症状依次为腹胀、腹水、双下肢水肿、乏力、纳差、黄疸、恶心和呕吐,少见症状为呕血、黑便,多数患者以多种症状合并出现。肝、脾肿大是最主要的体征,大多数患者肝功能异常。肝脏生化检查、影像学检查、临床表现、角膜色素环及铜代谢异常的生化指标,对HLD诊断和判断病情有重要意义。但该病临床表现多样,常缺乏特异性,首发症状不一,起病隐匿或慢性发展,容易误诊、漏诊,延误治疗,致残或致死率较高。早期诊断、及时治疗,可使患者症状缓解。本文回顾性分析20例HLD合并肝脏损害患者的临床误诊情况,旨在为该病的早期诊断提供参考。
1 资料与方法
1.1 临床资料
选取四川大学华西第四医院2012年1月至2014年7月HLD住院患者共20例,其中男12例,女8例,年龄4~41岁,平均17岁。
1.2 HLD诊断标准
按照Sternliep标准[1],具备以下两项者诊断为HLD:1)具有肝损害的临床表现;2)裂隙灯下检查角膜K-F环阳性;3)血清铜蓝蛋白降低;4)出现震颤、肌强直及面具样面容等神经系统表现。对于不典型病例的诊断可结合实验室的其他指标,如尿铜增加、血铜减低和游离血清铜升高,提示患者体内铜代谢异常,还可结合其阳性家族史。本组入选病例均符合以上标准且排除合并病毒性肝炎者。
2 结果
2.1 临床表现
20例HLD患者中,腹胀12例(60%),不同程度腹水12例(60%),双下肢水肿12例(60%),黄疸7例(35%),乏力、纳差7例(35%),仅体检发现肝功能异常、无明显临床症状6例(30%),肝区不适2例(10%),恶心、呕吐2例(10%),腹泻2例(10%),皮下出血1例(5%),消化不良1例(5%),无呕血,黑便。前两种表现居多,大部分患者合并多种表现(表1)。
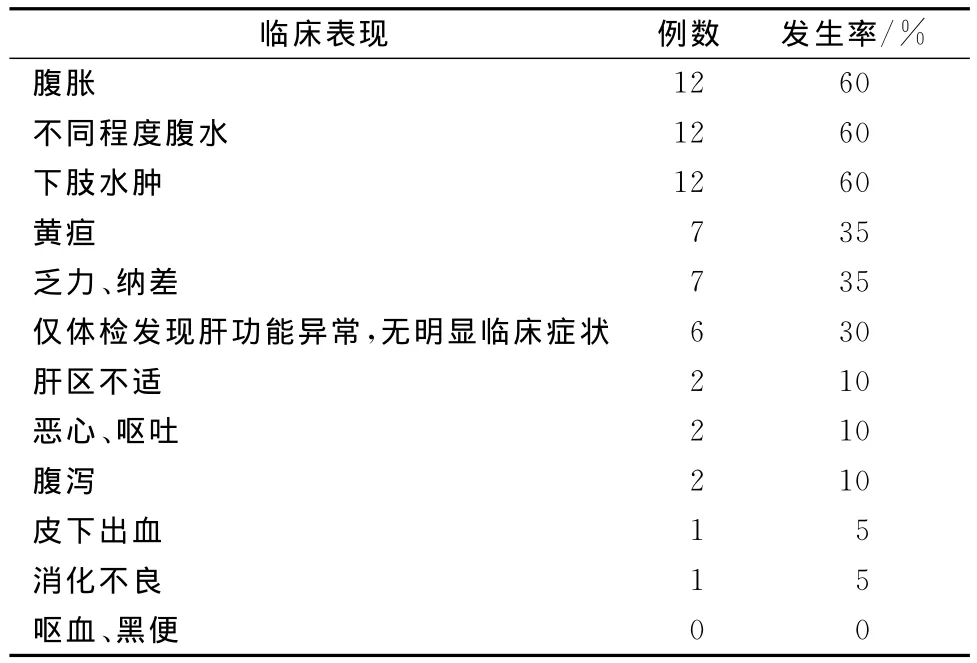
表1 20例HLD患者肝脏损害的临床表现
2.2 实验室及影像学检查
20例HLD患者实验室检查中,血清铜蓝蛋白、尿铜、谷草转氨酶、谷丙转氨酶和碱性磷酸酶阳性率较高(表2)。影像学检查结果见表3。
2.3 误诊情况
本组患者多以肝脏损害为主要表现,临床症状不典型,分别被误诊为慢性肝炎2例、肝炎后肝硬化代偿期6例及肝硬化失代偿期11例、自身免疫性肝病1例,在仅对症处理后,疗效不理想,经过多家医院诊治,最终确诊。误诊时间最短20d,最长5年,平均15.95个月。
2.4 治疗及预后
经严格低铜饮食,给予二巯基丙磺酸钠、青霉胺、硫酸锌等药物驱铜,复方二氯醋酸二异丙胺、还原型谷胱甘肽、复方甘草酸苷、多烯磷脂酰胆碱等药物保肝,补充多种维生素、钙剂,以及对症支持治疗后,20例患者均好转出院。
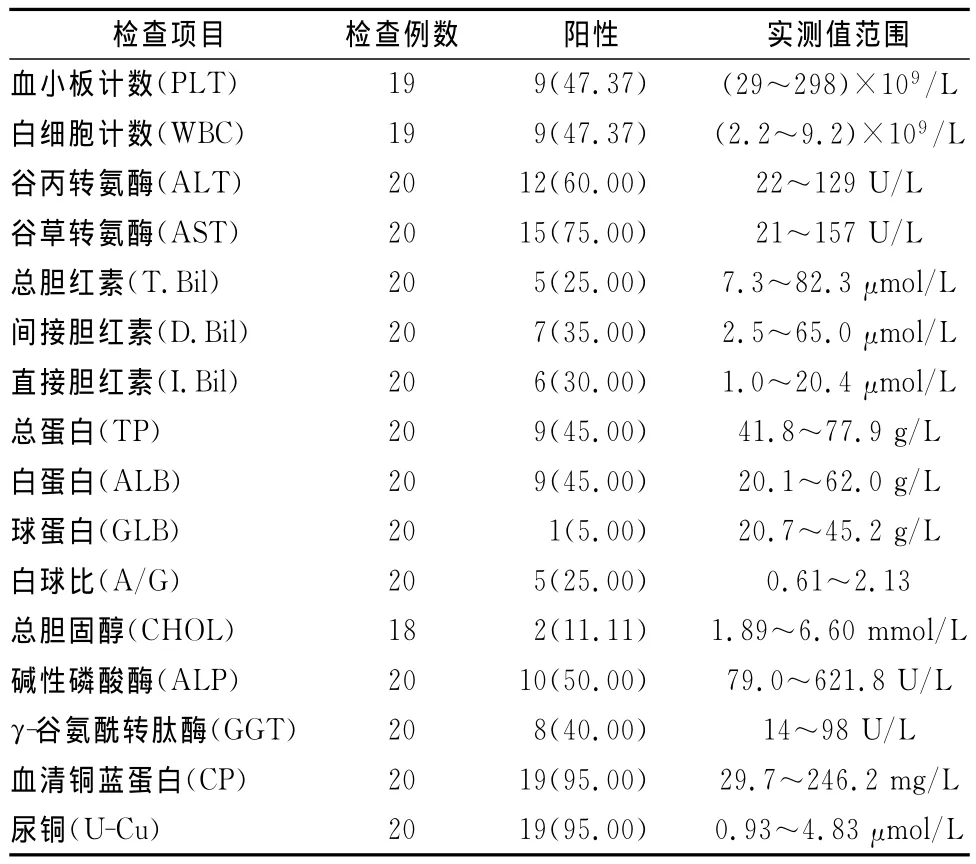
表2 20例HLD患者实验室检查结果[n(%)]

表3 部分HLD患者影像学检查阳性结果[n(%)]
3 讨论
HLD为一种常染色体隐性遗传病,位于13号染色体长臂的ATP7B基因突变是其发病的主要分子机制[2]。ATP7B基因主要在肝细胞表达,其编码的P型铜转运ATP酶位于肝细胞高尔基体外侧,功能是转运肝细胞内的铜,合成铜蓝蛋白并将铜排入胆汁。ATP7B基因突变可引发铜蓝蛋白合成障碍及胆道排铜障碍,导致过量的铜沉积在肝脏、大脑及角膜等全身各处,引发相应组织器官病变[2-4]。HLD虽属少见病,世界范围发病率为1/10万~1/3万[5],致病基因携带者比例约为 1/90[6-7]。我国该病发病率较高,且呈上升趋势。该病临床表现差异大,部分起病隐匿,表现复杂多样,发病初期肝功损害轻微,当其他系统症状出现时,多数已有肝硬化或门静脉高压。既往误诊的报道亦不少,但迄今为止其误诊率仍居高不下,尤其以肝脏损害为主要表现或首发症状的非脑症状起病者,更易被误诊为急慢性肝炎、肝硬化以及自身免疫性肝病等,致残率及致死率均较高。因此,在医疗活动中,医务工作者应开阔诊断思维,不能仅局限于临床常见病,在无常见病因可寻的情况下,应考虑本病的可能。早期发现、明确诊断,并给予正规、及时、准确的治疗,对患者的病情控制、疾病预后和转归,有着积极的意义。
本组患者多以肝脏损害为主要表现,临床症状不典型,分别被误诊为慢性肝炎2例、肝炎后肝硬化代偿期6例及肝硬化失代偿期11例、自身免疫性肝病1例。误诊时间最短20d,最长5年,平均15.95个月。分析临床误诊原因:1)本病起病隐匿,多数进展缓慢,早期症状多样化且不典型,多脏器受累,临床表现复杂,缺乏特异性症状,易与其他疾病混淆;2)首发症状不一,多数以肝病症状为首发,也有以贫血、消化道症状首发,部分亦合并神经、精神症状以及肾脏症状等;3)临床医师对该病认识不足,警惕性不高,临床思维局限于常见肝脏疾病;4)病史资料采集不够详尽,可能遗漏一些重要信息,忽视了血清铜、血清铜蓝蛋白、双眼K-F环等特异性检查;5)相应系统影像学表现不典型。
HLD是先天性遗传病中为数不多的可治疗疾病之一,早期诊断,及时恰当的治疗是改善预后的关键。要避免HLD合并肝脏损害的临床误诊,临床医师应提高对该病的认识,对下述患者应高度怀疑该病:不明原因的转氨酶升高、肝脾肿大、肝硬化、一过性黄疸、反复鼻及牙龈出血的患者;不明原因且治疗效果欠佳的肝病,特别是儿童及表面抗原阴性的患者;在肝病病史基础上合并精神、神经症状的患者;仅体检发现肝病体征,但家族成员中有该病,尤其是同胞一代有发病的患者。此外,应进一步检查角膜K-F环、血清铜蓝蛋白、铜氧化酶、尿铜、头颅MRI、腹部超声以及肝活检,以明确诊断,避免误诊。
[1]Sternliep I.Perspectives on Wilson′s disease[J].Hepatology,1990,12(5):1234-1239.
[2]European Association for Study of Liver.EASL Clinical Practice Guidelines:Wilson′s disease[J].J Hepatol,2012,56(3):671-685.
[3]Schilsky ML.Wilson disease:new insights into pathogenesis,diagnosis,and future therapy[J].Curr Gastroenterol Rep,2005,7(1):26-31.
[4]Tao TY,Gitlin JD.patic copper metabolism:insights from genetic disease[J].Hepatology,2003,37(6):124l-l247.
[5]Ala A,Walker AP,Ashkan K,et al.Wilson′s disease[J].Lancet,2007,369(9559):397-408.
[6]Figus A,Angius A,Loudianos G,et al.Molecular pathology and haplotype analysis of Wilson disease in Mediterranean population[J].Am J Hum Genet,1995,57(6):1318-1324.
[7]梁秀龄.解读《肝豆状核变性的诊断与治疗指南》[J].中国现代神经疾病杂志,2009,9(3):212-215.
