结合多组学和人类蛋白互作网络研究急性髓细胞白血病中的突变基因互作子网络
2015-11-19江建平窦同海
江建平,杨 芳,张 亮,窦同海,周 雁
(1.复旦大学 生命科学学院 微生物学与微生物工程系,上海 200438;2.上海人类基因组研究中心 上海市疾病与健康基因组学省部共建重点实验室,上海 201203)
急性髓细胞白血病(Acute Myelocytic Leukemia,AML)又称为急性非淋巴细胞白血病(ANLL),它包括所有非淋巴细胞来源的急性白血病.AML是一类造血系统的克隆性恶性疾病,它是由于多能干细胞或已轻度分化的前体细胞核型发生突变所形成.AML是一个具有高度异质性的疾病群,它可以由正常髓细胞分化发育过程中不同阶段的造血祖细胞恶性转化而来,并且起源于不同阶段祖细胞的AML 具有不同的生物学特征[1].AML在发达国家的发病率要高于发展中国家,西方国家高于东方国家,世界各地年发病率约为2.25/10万.随年龄增加,AML 发病率增高,在美国,30岁以下发病率为1.2/10万、80岁以上则高于20/10万[2].因此,AML 实际上是一种中、老年疾病.急性髓细胞白血病占成人急性白血病的80%~90%,占儿童急性白血病的15%~20%,男性的发病率高于女性.研究表明,在人群接受大剂量放射线或长期接触苯的情况下,AML发病率会增加[3].
研究表明,AML病人染色体结构发生变异,并且通过对染色体结构变异的识别,已经形成一些诊断标记用于临床诊断.但是,仍然有50%左右的病人在用检测染色体核型的方式进行诊断时,无法被确诊[4].通过靶基因定向测序和第二代测序(Next Generation Sequencing)技术已经确定了在AML 病人的基因组中常见的突变基因,包括:FLT3,NPM1,KIT,CEBPA,TET2,DNMT3A 以及IDH1[5-8].然而近期的研究表明,有些AML病人的这些关联基因中并未发现任何突变[9],所以可能存在新的AML 关联基因,有必要对AML病人基因组中所有的突变基因进行更加深入的信息挖掘.
基于第二代测序技术的全基因组测序(Whole Genome Sequencing,WGS),全外显子组测序(Whole Exome Sequencing,WES)以及转录组测序(Transcriptome Sequencing)极大地加速了疾病发病机制的研究.2008年,Ley等发表了1例AML患者的全基因测序结果,该研究发现了8个体细胞突变,对初发和复发肿瘤样本的突变区域进行扩增后发现,FLT3-ITD 和NPM1是疾病发展过程中相关的基因,但是据初发和复发肿瘤样本中FLT3-ITD 出现率推断出FLT3-ITD 并不存在于所有肿瘤细胞中,FLT3-ITD 的突变可能发生于肿瘤病程的后期[10].
生命科学研究已经进入后基因组时代,各大公共数据库中已经积累大量的基于不同技术的组学数据,其中包含有多个研究机构研究并共享的数据,如人类基因组计划(Human Genome Project,HGP),1 000人基因组计划(1 000Genomes),癌症和肿瘤基因图谱计划(The Cancer Genome Atlas,TCGA)数据库等.除了大规模研究机构共享的数据以外,还有大量由个人或研究机构提交到美国国立生物信息中心(NCBI)数据库中共享的数据,如NCBI基因组(Genome)数据库,基因表达组(GEO)数据库,基因型和表型(dbGAP)数据库等.这些海量数据虽然方便了研究者们将已有数据直接应用于自己的研究中,但如何有效地对不同类型的组学数据进行综合解读成为了一大难题.Califano等提出使用数据整合的方式来研究生物学机制,他们试图将全基因组关联分析(Genome Wide Association Study,GWAS)数据和基因调控网络数据进行整合,使GWAS所获得的差异基因在基因调控网络中进行富集,并获得富集的调控网络,再对网络进行研究[11].Han 等使用整合全基因组关联分析和人类蛋白互作网络(Human Protein Interaction Network,HPIN)的方法研究欧洲人的酒精依赖症,并且发现了一些新的可能增加患酒精依赖症风险的代谢通路以及基因[12].许多研究均表明,结合不同组学数据进行疾病的研究可以有效地发现单一组学数据研究中无法发现的疾病相关基因,并且不同组学数据之间可以进行互相补充,从而更加准确地解释疾病的发病机制.
本文中结合AML病人的基因组中基因突变数据,基于RNA-seq的转录组表达量数据以及人类蛋白互作网络数据,通过构建网络的方式,研究基因组中突变基因的功能,并试图发现突变基因所富集的网络,从而为AML发病机制的阐明和诊断提供一些依据.
1 材料和方法
1.1 数据描述
AML基因组体细胞突变数据和RNA 转录组数据来源于TCGA 数据库(https:∥tcga-data.ncbi.nih.gov/tcga/),数据集中包括200例病人的病变组织样本.在TCGA 的初步分析数据中,200个样本包含有体细胞突变数据,173个样本包含有RNA 转录组的表达量数据[13].本文筛选出同时包含体细胞突变数据和RNA 转录组数据的样本进行分析,筛选获得173例AML 数据,其中153个白人样本,13个黑人样本,以及7个其他人种样本.样本中92个为男性,81个为女性.
1.2 基因集筛选
使用UCSC Genomes(http:∥genome.ucsc.edu/)数据库对AML体细胞突变数据中的基因进行名称转换,将数据中的原始基因名转换为基于HCNC(Human Gene Nomenclature)的标准基因名.去除无法被转换为HCNC基因名的基因,筛选并去除在所有研究样本中均不表达的基因(RNA转录组表达量为0).
1.3 统计学分析
1.3.1 基因突变频率的二项检验
使用二项检验对AML病人的基因突变频率进行检验,其中基因期望突变频率是单位长度突变次数乘以基因的最长转录本长度.参考Lawrence等[14]对癌症基因组中基因突变率的研究,本文使用单位长度突变次数为0.4次/Mb.基因转录本长度信息来源于UCSC Genomes数据库,基因观察到的突变率为基因在所有样本中的突变次数除以样本总数.使用R 统计学软件对每个基因进行二项检验,检验时使用单尾检验,并计算P 值,其中零假设为观察到的基因突变频率显著不高于AML 病人基因组中基因的平均突变频率.
1.3.2 高突变率基因互作网络分析
使用R 软件包dmGWAS[15]结合突变基因的RNA 转录组数据和HPIN 对AML 病人基因组中突变基因进行分析.dmGWAS 分析中使用的基因权值为二项检验所获得的P 值,互作网络数据来源于PINA[16],使用USCS Genomes数据库将蛋白质互作网络中Uniprot基因名转化为标准的HCNC 基因名,最终获得约16.7万对蛋白质互作数据.dmGWAS软件包以HPIN 为基础,通过计算模块值(S)的方式进行模块的识别和拓展,S 的计算结合边权重(基因间RNA 表达量的Pearson相关系数)和节点权重(基因二项检验的P 值),S 的计算公式[15]如下:

其中λ是权衡边权重和节点权重的参数,本文中使用程序估算获得的λ,新增加基因后,如果S 值的增量小于r×S 时,dmGWAS停止模块拓展,本文中使用的r值为0.1.计算获得S 值后,根据模块的大小对S值进行修正,使用的修正公式为Sn=(S-μ)/σ.其中μ 和σ 分别为根据模块大小随机抽样10 000次获得的均值和方差;根据Sn大小对模块进行降序排列.一般认为,Sn值越大的模块与疾病的相关性越高,所以通常选取Sn靠前的模块构建子网络.
1.3.3 基因富集分析
使用在线分析工具DAVID[17]分别对候选基因集、高突变率基因(P<0.05)和子网络中基因进行GO[18]和KEGG[19]代谢通路富集分析.使用DAVID 中的Fisher精确检验对富集的GO 和KEGG 代谢通路进行统计检验并计算P 值,使用Bonferroni对多重检验的P 值进行校正.
2 结果
2.1 候选基因集
根据体细胞突变数据和RNA 转录组数据对样本进行筛选,得到173例AML 病例数据.使用USCS Genomes数据库将数据中基因名统一转换为HCNC基因名,其中无法识别基因名的基因被去除,筛选并去除在173例样本中均不表达的基因,最终得到1 450个基因满足以上条件,并用于后续分析.
对173例病人的1 450个基因统计后发现,AML病人基因组中22条常染色体和X 染色(23号)上均发现了体细胞突变基因。其中1号染色体包含有最多体细胞突变基因,为130个基因,而18号,21号染色体相对于其他染色体,包含较少的体细胞突变基因,分别为17个和18个体细胞突变基因.
2.2 候选基因集KEGG 代谢通路和GO 富集分析
通过对1 450个体细胞突变基因进行KEGG 的代谢通路富集分析后,发现15个代谢通路在1 450个体细胞突变基因中发生了富集(P<0.05)(表1).经过Bonferroni方法进行多重检验的P 值校正后,仍有1个代谢通路,黏着斑通路(hsa04510)的P 值达到显著水平.在富集的15个代谢通路中,有4个是与癌症直接相关的,包括神经胶质瘤通路(hsa05214),慢性髓细胞白血病通路(hsa05220),癌症代谢通路(hsa05200)和急性髓细胞白血病通路(hsa05221);还有一些与其他疾病相关的代谢通路,如扩张型心肌病通路(hsa05414),Ⅱ型糖尿病通路(hsa04930)等.
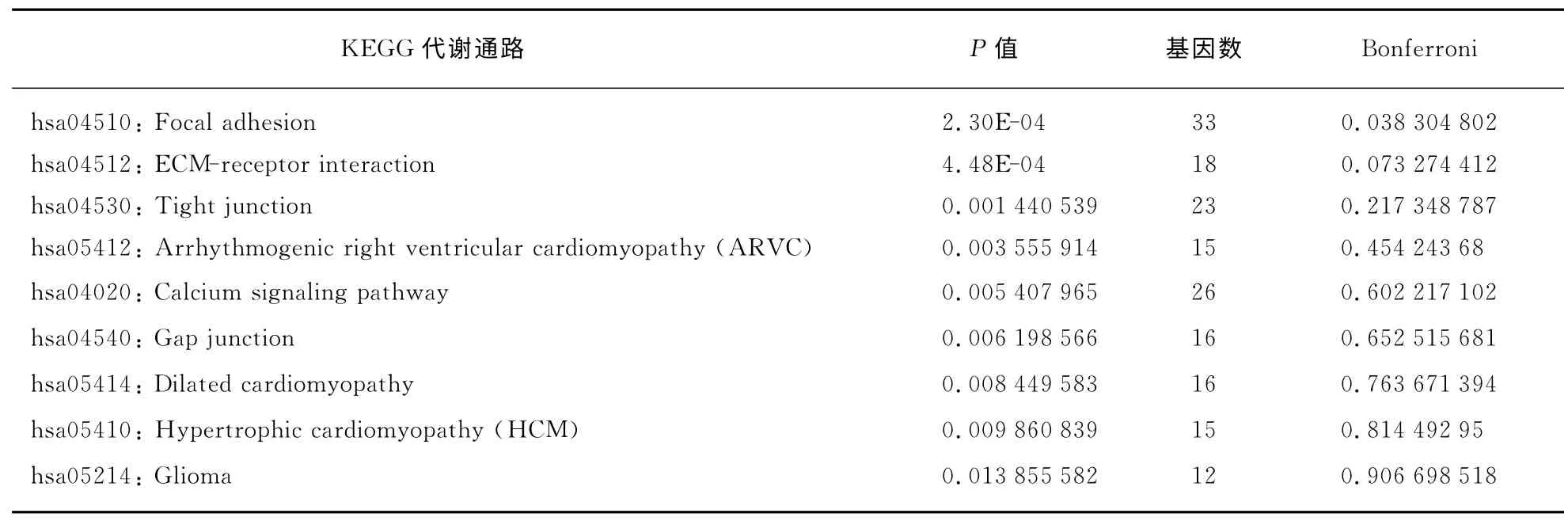
表1 AML中体细胞突变基因在KEGG 代谢通路富集分析中富集的通路Tab.1 The enriched pathways from KEGG pathway analysis of somatic mutated genes in AML
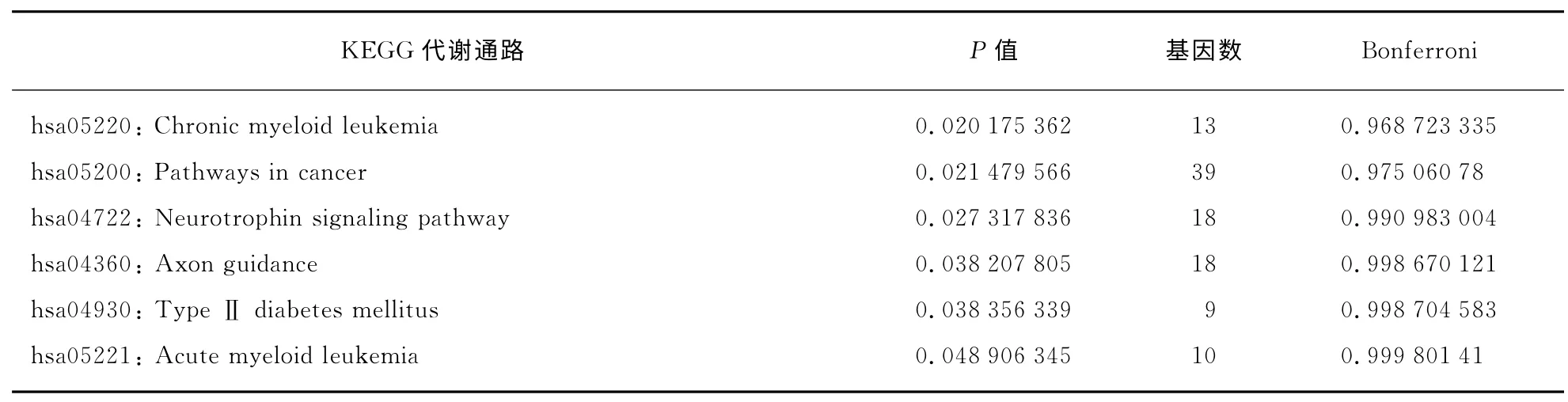
(续表)
使用DAVID 在线工具对1 450个体细胞突变基因进行GO 富集分析,富集所得的GO 三大分类(生物过程、细胞组分和分子功能)中的前5位结果如下表(表2).从GO 三大类的富集结果中可以看出,在生物过程中,体细胞突变基因在细胞粘着的过程发生富集;细胞组分中,染色体组成相关的组分发生富集;分子功能中,离子通道和跨膜转运的功能发生富集.

表2 候选基因集在三大类GO 富集分析中富集的前5个GO 注释Tab.2 The top 5items in three main categories from GO enrichment analysis of candidate genes
2.3 基因突变频率的二项检验
使用二项检验对173例样本中的1 450个候选基因进行检验,从结果(图1)中可以看出,大多数体细胞突变基因的P 值不显著,只有约8%(114/1 450)的体细胞突变基因P 值达到统计学显著水平(P<0.05),其中5号,13号和2号染色体分别包含有AML病人基因组中P 值最为显著的3个体细胞突变基因(NPM1,FLT3和DNMT3A).在18号染色体的17个体细胞突变基因中,未发现差异显著的体细胞突变基因;在7号染色体上,差异显著的体细胞突变基因占其染色体上总的体细胞突变基因的比例最多,为14.5%(10/69);21号染色体尽管整体包含较少的体细胞突变基因,但是其包含的2个P 值较显著的体细胞突变基因.部分染色体上虽然包含有较多的体细胞突变基因,但是包含P 值显著的体细胞突变基因较少,如6号染色体.
表3中列出了20个P 值较小的体细胞突变基因,其中NPM1,FLT3和DNMT3A 分别在48、49和48例样本中被检测到体细胞突变,11个高突变率基因在大于10例样本中被检测出突变.

图1 1 450个体细胞突变基因二项检验的P 值分布(彩页见封3)Fig.1 The distribution of Pvalue in the binomial test of 1 450somatic mutated genes

图1 1 450个体细胞突变基因二项检验的P 值分布Fig.1 The distribution of Pvalue in the binomial test of 1 450somatic mutated genes

表3 二项检验中P 值较小的前20个基因及其相关信息Tab.3 The general features of top 20low Pvalue genes in the binomial test
2.4 高突变频率基因集KEGG 代谢通路和GO 富集分析
通过对二项检验结果中基因突变频率显著高于平均突变频率的114个体细胞突变基因进行KEGG的代谢通路富集分析后,发现5个代谢通路在114个体细胞突变基因中发生了富集(P<0.05)(表4).经过Bonferroni方法进行多重检验的P 值校正后,仍有1个代谢通路,即急性髓细胞白血病通路(hsa05221)P 值达到显著水平(<0.05).在5个富集的代谢通路中,2个是与白血病直接相关的代谢通路(hsa05221和hsa05220),1个是甲状腺癌相关代谢通路(hsa05216).

表4 高突变频率基因集在KEGG 代谢通路富集分析中富集的通路Tab.4 The enriched pathways from KEGG pathway analysis of frequently mutated genes
使用DAVID 在线工具对114个高突变频率基因进行GO 富集分析,富集所得的GO 三大分类(生物过程、细胞组分和分子功能)中的前5位结果如下表(表5).从GO 三大类的富集结果中可以看出,在生物过程中,高突变频率基因在神经突触调控和白细胞分化的过程发生富集;细胞组分中,染色体组成相关的组分发生富集;分子功能中、染色体结合,通道激活和跨膜转运的功能发生富集.

表5 高突变频率基因集在三大类GO 富集分析中富集的前5个GO 注释Tab.5 The top 5items in three main categories from GO enrichment analysis of frequently mutated genes
2.5 互作子网络分析
使用R 软件包dmGWAS将1 450个体细胞突变基因结合RNA 转录组数据和蛋白互作网络数据进行网络分析,获得288个基因互作功能模块.为了获得与疾病相关性较高的互作子网络,本文选取前5%模块值较大的功能模块构建基因互作子网络(图2),其中包含有21个体细胞突变基因.互作子网络中节点表示基因,连线表示基因间的相互作用,节点颜色的深浅表示节点的权重,颜色越浅权重值越大(P 值越小),连线的粗细表示节点间的相关性,线条越粗,相关性约高.
2.6 互作子网络中基因的KEGG 代谢通路和GO 富集分析
使用DAVID 在线工具对互作子网络中的21个体细胞突变基因进行KEGG 代谢通路的富集分析,分析结果如下表(表6).其中3条代谢通路在21个体细胞突变基因中富集(P<0.05),分别为细胞循环(hsa04110),慢性髓细胞白血病(hsa05220)和趋化因子信号通路(hsa04062),经过Bonferroni方法进行多重检验的P 值校正后,细胞循环相关的代谢通路的P 达到显著水平.


图2 AML中体细胞突变基因的互作子网络Fig.2 The interaction subnetwork of somatic mutated genes in AML

表6 互作子网络中基因在KEGG 代谢通路富集分析中富集的通路Tab.6 The enriched pathways from KEGG pathway analysis of genes from subnetwork
使用DAVID 在线工具对互作子网络中21个体细胞突变基因进行GO 富集分析,富集所得的GO 三大分类(生物过程,细胞组分和分子功能)中的前5位结果如表7所示.从GO 三大类的富集结果中可以看出,在生物过程中,子网络中的基因在染色体组成和修饰过程发生富集;细胞组分中,染色体组成相关的组分发生富集;分子功能中,染色体结合,转录调控和转录因子结合的功能发生富集.
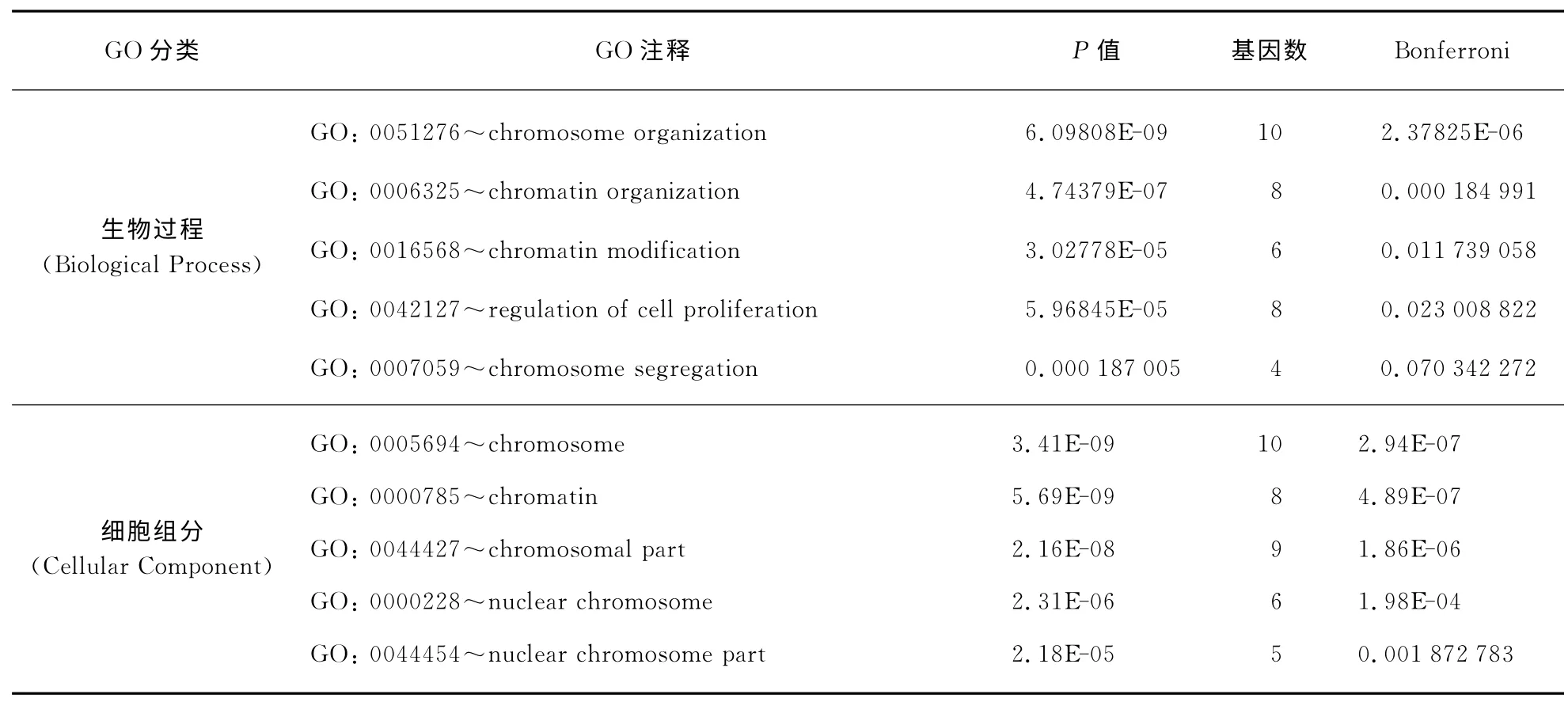
表7 互作子网络中基因在三大类GO 富集分析中富集的前5个GO 注释Tab.7 The top 5items in three main categories from GO enrichment analysis of genes from subnetwork

(续表)
3 讨论
本文首先对1 450个AML中体细胞突变基因进行KEGG 代谢通路和GO 富集分析.GO 富集分析结果表明,在生物过程中,体细胞突变基因在细胞黏着的过程发生富集;细胞组分中,染色体组成相关的组分发生富集;分子功能中,离子通道和跨膜转运的功能发生富集.已有研究表明,细胞黏着在很多恶性疾病的发展过程中具有重要作用,对细胞黏着过程中分子的相互作用进行调控,是一种治疗AML 的有效手段[20].可以看出,单纯对AML中体细胞突变基因的富集分析,发现一些与AML 相关的生物过程发生了异常.
使用二项检验对173例AML病人基因组中1 450个体细胞突变基因进行检验,发现在AML病人基因组中常见的突变基因的突变频率均显著高于基因组中基因的平均突变频率,这一结果表明FLT3,NPM1,KIT,CEBPA,TET2,DNMT3A 以及IDH1基因在这173例AML 病人中也是较为常见的突变基因.以P 值小于0.05作为差异显著的标准,经二项检验后,获得了114个突变频率显著高于平均突变频率的基因,对这114个突变频率较高的基因进行KEGG 代谢通路和GO 的富集分析,KEGG 代谢通路富集结果表明,114个突变基因在急性髓细胞白血病(hsa05221)和慢性髓细胞白血病(hsa05220)代谢通路中发生了富集,这两个代谢通路都是与白血病直接相关的疾病代谢通路,KEGG 代谢通路富集结果一方面说明白血病代谢通路中的基因突变频率显著高于其他代谢通路,另一方面说明,AML 发病机制相关的基因突变可能会包含在突变频率较高的基因集中.GO 富集结果表明,在生物过程中,高突变频率基因在神经突触调控和白细胞分化的过程发生富集;细胞组分中,染色体组成相关的组分发生富集;分子功能中,染色体结合,通道激活和跨膜转运的功能发生富集.这些AML 病人的神经系统或相关功能可能出现异常;中枢神经系统损伤是急性白血病的常见并发症之一,许多AML 的中枢神经系统会受到损伤[21],高突变率基因的GO 富集结果说明AML病人的基因组中,许多中枢神经系统功能相关的基因发生突变可能是AML 的中枢神经系统发生损伤的主要原因.虽然使用对基因富集的方法可以发现AML 中某些功能发生异常,但是仍然无法揭示这些基因突变的产生是AML作用的结果还是导致AML 的原因,所以需要我们从基因间相互作用的角度去研究AML中的突变基因.
本文使用AML中1 450个体细胞突变基因结合人类蛋白互作数据和基因的RNA 转录组数据,构建基因互作子网络,互作子网络中包含有21个体细胞突变基因,其中部分基因是AML中常见的突变基因,如NPM1,SMC3等,另一些突变基因则在AML 中出现的频率较低,如HIST3 H3,RBBP4等,但这些基因与AML中常见突变基因间具有相互作用;互作子网络中包含5个驱动基因(相邻基因数大于5):SMC3,HDAC2,NPM1,RBBP4和HIST3 H3,这些驱动基因可能与AML 的致病机制密切相关;其中NPM1是已知的与AML 相关的基因[13];SMC3是黏连蛋白基因,在有丝分裂的过程中使得姐妹染色单体粘连在一起,该基因的突变可能会导致染色体核型的改变,并且SMC3的突变与NPM1之间具有很强的相关性[22];HDAC 是一类组蛋白脱乙酰酶基因,对HDAC 进行阻断是临床上治疗癌症的有效手段[23];HIST3 H3是组蛋白基因,主要是控制细胞核中染色体牵丝的形成,HIST3 H3的突变可能会导致染色体核型发生改变,在AML 中对HIST3 H3 研究较少,而对AML 中HIST3 H3 的深入研究,可能会揭示AML病人基因组中染色体核型变化的原因;RBBP4是与组蛋白乙酰化和染色体组装相关的基因,通过对RBBP4表达的上下调可以控制细胞的循环和凋亡[24].使用DAVID 在线工具对21个基因进行KEGG代谢通路富集分析后发现,3条代谢通路在21个基因中发生了富集,其中趋化因子代谢通路与AML 的致病机制相关性的研究较少,趋化因子在AML中细胞凋亡过程具有重要的调控作用,Kremer等的研究表明,趋化因子受体SDF-1/CXCR4可以诱导急性髓细胞白血病中细胞凋亡[25],趋化因子信号通路基因发生突变,可能会导致白细胞细胞凋亡过程异常,从而使得白细胞发生恶性增殖.互作子网络中的体细胞突变基因代谢通路富集结果表明,趋化因子信号通路与急性髓细胞白血病的发病机制有密切关系.本文中还发现了一些AML潜在的相关基因,如CBX7,GNAI2和COPS2等;已有研究[26]表明,在甲状腺癌中,CBX7表达水平发生下调,并且GBX7的表达水平与甲状腺癌的恶性程度相关;在AML 中,CBX7突变可能引起GBX7表达水平变化,从而影响AML的发生或恶性程度.
综上,使用不同的研究手段可以发现基因突变与疾病间的相互关系,并且,将基因组中发生突变的基因结合基因的RNA 转录组和人类蛋白互作网络,可以发现某些疾病基因组中突变基因间互作的子网络,从而发现那些在传统研究方法中被忽视的基因或代谢通路;本文使用基于HPIN 网络的方法研究突变基因,可以有效的揭示疾病中潜在的驱动基因,这些驱动基因可能在AML 的发病机制中具有重要作用,对这些驱动基因的进一步研究,可能会发现新的AML发病相关基因,为将来AML 的诊断以及个性化医疗提供一定的依据.
[1]王福旭.造血干细胞移植治疗高危急性髓细胞白血病的研究进展[J].国际输血及血液学杂志,2013,36(5):412-417.
[2]羊裔明.急性髓细胞白血病治疗现状及其展望[J].临床误诊误治,2006,19(9):1-4.
[3]王 星,王小钦,顾静文,等.成人急性髓细胞白血病发病危险因素的研究[J].中华全科医师杂志,2011,10(9):637-640.
[4]Walter M J,Payton J E,Ries R E,et al.Acquired copy number alterations in adult acute myeloid leukemia genomes[J].Proc Natl Acad Sci USA,2009,106(31):12950-12955.
[5]Stirewalt D L,Radich J P.The role of FLT3in haematopoietic malignancies[J].Nature Reviews Cancer,2003,3(9):650-665.
[6]Bacher U,Schnittger S,Haferlach T.Molecular genetics in acute myeloid leukemia[J].Current Opinion in Oncology,2010,22(6):646-655.
[7]Ley T J,Ding L,Walter M J,et al.DNMT3A mutations in acute myeloid leukemia[J].The New England Journal of Medicine,2010,363(25):2424-2433.
[8]Yamashita Y,Yuan J,Suetake I,et al.Array-based genomic resequencing of human leukemia[J].Oncogene,2010,29(25):3723-3731.
[9]Shen Y,Zhu Y M,Fan X,et al.Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia[J].Blood,2011,118(20):5593-5603.
[10]Ley T J,Mardis E R,Ding L,et al.DNA sequencing of a cytogenetically normal acute myeloid leukemia genome[J].Nature,2008,456(7218):66-72.
[11]Califano A,Butte A J,Friend S,et al.Leveraging models of cell regulation and GWAS data in integrative network-based association studies[J].Nat Genet,2012,44(8):841-847.
[12]Han S,Yang B Z,Kranzler H R,et al.Integrating GWASs and human protein interaction networks identifies a gene subnetwork underlying alcohol dependence[J].American Journal of Human Genetics,2013,93(6):1027-1034.
[13]The Cancer Genome Atlas Research Network.Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia[J].The New England Journal of Medicine,2013,368(22):2059-2074.
[14]Lawrence M S,Stojanov P,Mermel C H,et al.Discovery and saturation analysis of cancer genes across 21tumour types[J].Nature,2014,505(7484):495-501.
[15]Jia P,Zheng S,Long J,et al.dmGWAS:Dense module searching for genome-wide association studies in protein-protein interaction networks[J].Bioinformatics,2011,27(1):95-102.
[16]Wu J,Vallenius T,Ovaska K,et al.Integrated network analysis platform for protein-protein interactions[J].Nat Methods,2009,6(1):75-77.
[17]Huang Da W,Sherman B T,Lempicki R A.Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources[J].Nature Protocols,2008,4(1):44-57.
[18]Ashburner M,Ball C A,Blake J A,et al.Gene ontology:Tool for the unification of biology.The Gene Ontology Consortium[J].Nat Genet,2000,25(1):25-29.
[19]Du J,Yuan Z,Ma Z,et al.KEGG-PATH:Kyoto encyclopedia of genes and genomes-based pathway analysis using apath analysis model[J].Molecular Biosystems,2014,10(9):2441-2447.
[20]Kupsa T,Horacek J M,Jebavy L.The role of adhesion molecules in acute myeloid leukemia and(hemato)oncology:A systematic review[M].Olomouc,Czechoslovakia:Biomedical papers of the Medical Faculty of the University Palacky,2014.
[21]周小平,张永宁.3例急性白血病并发中枢神经系统损害患者的临床特征分析及文献复习[J].中国临床神经科学,2013,21(4):418-422,437.
[22]Thol F,Bollin R,Gehlhaar M,et al.Mutations in the cohesin complex in acute myeloid leukemia:Clinical and prognostic implications[J].Blood,2014,123(6):914-920.
[23]Boissinot M,Inman M,Hempshall A,et al.Induction of differentiation and apoptosis in leukaemic cell lines by the novel benzamide family histone deacetylase 2and 3inhibitor MI-192[J].Leukemia Research,2012,36(10):1304-1310.
[24]Casas S,Ollila J,Aventin A,et al.Changes in apoptosis-related pathways in acute myelocytic leukemia[J].Cancer Genetics and Cytogenetics,2003,146(2):89-101.
[25]Kremer K N,Peterson K L,Schneider P A,et al.CXCR4 chemokine receptor signaling induces apoptosis in acute myeloid leukemia cells via regulation of the Bcl-2family members Bcl-XL,Noxa,and Bak[J].The Journal of Biological Chemistry,2013,288(32):22899-22914.
[26]Pallante P,Federico A,Berlingieri M T,et al.Loss of the CBX7gene expression correlates with a highly malignant phenotype in thyroid cancer[J].Cancer Research,2008,68(16):6770-6778.
