p38-p53-p21信号通路参与调控血管紧张素-Ⅱ诱导的人血管平滑肌细胞衰老
2015-11-19廉亚茹刘新华潘礼龙胡金锋
廉亚茹 ,刘新华,韩 苗,潘礼龙,胡金锋,魏 刚,倪 挺
(1.复旦大学 生命科学学院,现代人类学教育部重点实验室,上海 200438;2.复旦大学 遗传工程国家重点实验室,上海 200438;3.复旦大学 药学院,上海 201203)
个体衰老是一个涉及心血管迁移率、死亡率升高且已知心血管因子增加的生理过程[1].个体衰老通常伴随着血管系统的改变,特别是动脉的结构和功能[2].在衰老的过程中,血管内皮细胞和平滑肌细胞发生改变.例如,在衰老过程中,内皮产生的血管舒张剂如一氧化氮(NO)或/和环前列腺素的降低以及血管平滑肌细胞对它们的响应能力的降低,导致内皮依赖性血管舒张受损[3].而且,在衰老的动脉中,促炎症因子和促血栓形成因子表达增加[4].值得注意的是,体外衰老的血管细胞也有类似的功能改变[5-8],这使得研究衰老过程有了较好的体外试验模型.
血管紧张素-Ⅱ(Angiotensin Ⅱ,Ang Ⅱ)是肾素-血管紧张素系统(Renin-Angiotensin System,RAS)中一个重要的生物活性肽[9],在血管的结构和功能调控方面有重要的作用.血管紧张素-Ⅱ是血管壁向全身或者局部分泌的多功能血管活性肽,广泛参与心血管疾病的发生,而血管平滑肌细胞是它一系列活动的重要靶标.Ang Ⅱ是一个强效的血管收缩剂,可以通过多个细胞信号通路的作用,促使血管细胞发生有丝分裂、炎症反应和纤维化.在病理条件下,Ang Ⅱ信号通路会导致内皮功能紊乱、血管重塑和炎症反应,并参与心血管疾病中血管损伤的重要过程.
研究显示,伴随着机体代谢形成的Ang Ⅱ激活后在体内外均可诱导血管平滑肌细胞发生衰老[10-12].此外,越来越多的证据表明,阻断AT1受体或者抑制血管紧张素转换酶能够抑制Ang Ⅱ引起的血管平滑肌细胞衰老,并且对抵抗衰老过程和衰老相关的血管疾病,例如高血压和动脉粥样硬化,有积极的作用[13-14].研究发现,抑制Ang Ⅱ活性能降低心血管疾病的发病率和死亡率[15].然而,在人的血管平滑肌细胞中,衰老的细胞在RNA 和蛋白质层面上到底发生了哪些改变,Ang Ⅱ通过何种信号通路引起细胞衰老,各种不同信号通路是否在其中起作用则还没有非常系统的回答.因此为了揭示Ang Ⅱ导致人的血管平滑肌细胞衰老的分子机制,论文通过转录组测序(RNA-seq),结合实时定量PCR(qPCR)以及Western blot方法的实验验证,发现参与其他细胞衰老的TGF-β信号通路在Ang Ⅱ诱导的人血管平滑肌细胞衰老中明显被抑制,p38-p53-p21信号通路参与了人血管平滑肌细胞的诱导衰老过程,而与之平行的p38-p16信号通路则不参与该调控过程.
1 材料和方法
1.1 细胞培养及RNA-seq文库构建
原代人血管平滑肌细胞(human Vascular Smooth Muscle Cell,hVSMC;ScienCell Research Laboratories,#6110)培养至P3代,用100nmol/L的AngⅡ分别刺激48h(48h)、72h(72h).刺激结束后一部分采用Senescence Cells Histochemical Staining Kit(Sigma-Aldrich)进行染色并采用Western blot进行衰老相关蛋白表达检测;另一部分加入1mL TRIzol试剂用枪头小心吹打混匀提取总RNA,构建链特异性RNA-seq文库(dUTP介导).构建的文库通过质量控制后在HiSeq2500平台上进行高通量测序(上海晶能生物技术公司).
1.2 RNA-seq数据分析
利用FastQC软件对RNAseq数据的测序质量进行评估;在UCSC(http:∥genome.ucsc.edu/)中下载人类基因组序列以及转录组注释文件;然后利用Tophat2[16]将RNAseq数据比对到人参考基因组上(hg19),其中参数-r和——mate-std-dev parameters表示的是测序片段中间距离的平均值和标准差,可以用Bowtie[17]软件进行估计;接下来用Cufflinks进行转录本的组装,利用Cuffmerge将组装的转录本进行合并,最后通过Cuffdiff进行差异表达基因的比较分析[18].所有参数没有特殊说明的均设为默认值.
1.3 实时定量PCR(qPCR)
将去除基因组DNA 污染的1μg总RNA 进行反转录组,引物采用Oligo-dTVN(5’-TTTTTTTT TTTTTTTTTTVN-3’,V 表示T 之外的3个碱基,而N 为任何4个碱基),逆转录酶购买自天根公司.使用primer 3.0在线设计qPCR 引物(表1),并在生工公司合成,然后采用天根公司的SYBR Green PCR kit实时荧光PCR 试剂盒进行qPCR 检测.退火温度根据不同引物序列作相应的调整,并通过熔解曲线判断扩增的特异性.

表1 qPCR 检测所用引物序列Tab.1 Primers used in qPCR
1.4 Western blot
p16抗体(R&D,AF5779),β-actin抗体(SANTA CRUZ,SC-47778),p38抗体(#4631),MKK3抗体(#5674)都来自Cell Signaling Technology,二抗(Jackson ImmunoResearch,107487).采用半干转膜的方法,按照各自抗体说明书进行抗体稀释,采用Tanon 5200 全自动化学发光图像分析系统(Tanon)对Western blot的结果进行分析.
2 结果
2.1 AngⅡ诱导人血管平滑肌细胞发生衰老
为了确定血管紧张素-Ⅱ(AngⅡ)是否会诱导人的血管平滑肌细胞(hVSMC)发生衰老,研究采用100nmol/L的AngⅡ分别刺激P3代(从原代数起第3代)的hVSMC细胞48h和72h.衰老相关的β-半乳糖苷酶活性是鉴定人细胞衰老的一个生物学标志物[19].实验发现,与对照组相比,处理组细胞β-半乳糖苷酶(β-Gal)染色明显增多(图1(a)),并且与48h处理相比,72h处理组中蓝染的细胞更多.而且,Western blot结果表明AngⅡ处理的细胞中,细胞衰老的重要分子标记物p53和p21的蛋白水平明显升高,并且随着处理时间延长,升高越明显(图1(b)).以上结果说明AngⅡ的刺激引起了人的血管平滑肌细胞发生衰老.

图1 Ang Ⅱ诱导人血管平滑肌细胞衰老Fig.1 AngⅡinduces hVSMC senescence
2.2 AngⅡ诱导hVSMC衰老的基因表达谱变化
为了更全面和系统地研究人原代平滑肌细胞在诱导型早衰中的基因表达变化规律,研究选取了AngⅡ诱导前、诱导后48h(Ang Ⅱ_48h)和诱导后72h(Ang Ⅱ_72h)3个时间点,构建dUTP介导的链特异性RNA-seq文库,结合高通量测序和生物信息学分析,探索在诱导的不同时间点,基因的差异表达情况.我们以变化幅度超过2倍,P≤0.05作为差异表达基因的标准.结果发现,相比于诱导前,共有628个基因和487个基因分别在Ang Ⅱ_48h和Ang Ⅱ_72h发生了差异表达.
为了探究哪些基因在不同时间处理时均有差异表达,我们对这两个时间点差异表达的基因进行了重叠分析,发现有210个基因在48h和72h处理时均差异表达,这一基因数目与在Ang Ⅱ_48h处理和Ang Ⅱ_72h处理时特异调控的基因数目相比都要小.
我们对于在Ang Ⅱ_48h 和Ang Ⅱ_72h 处理时均差异表达的210 个基因进行了GO(Gene Ontology)生物学功能富集分析(表2).结果显示,在富集的前10个GO 生物学功能中都是与刺激响应和VSMC衰老相关的,例如转录调控、代谢调控、细胞黏附等,这进一步说明48h和72h的Ang Ⅱ处理确实引起了hVSMC发生了细胞衰老[23].
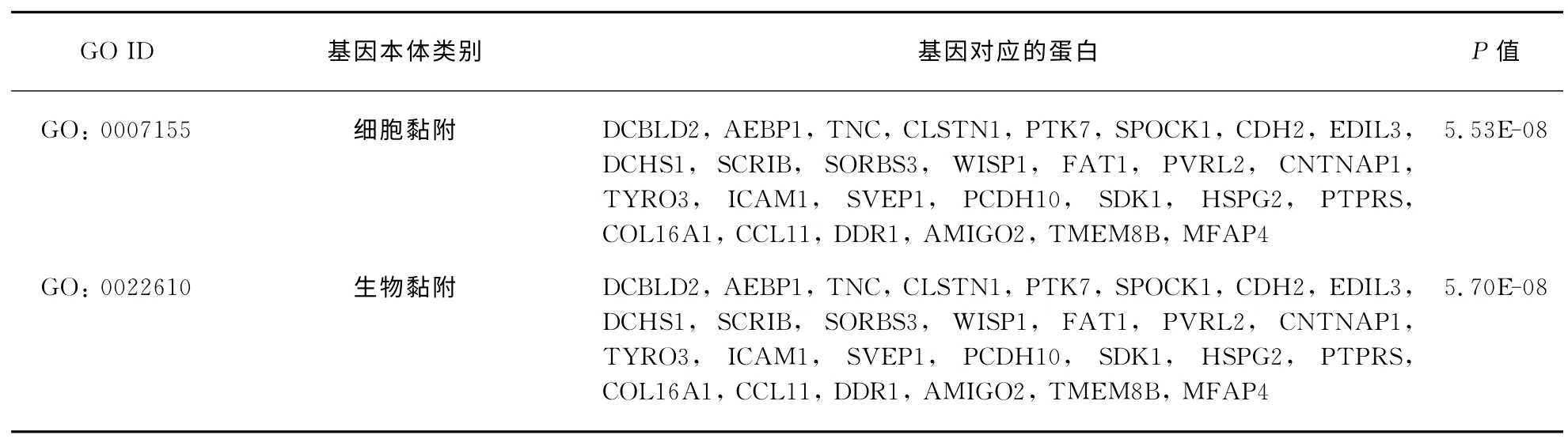
表2 AngⅡ处理48h&72h均差异表达的基因GO 生物学功能富集分析Tab.2 GO-analysis of both different expression genes induced by Ang Ⅱfor 48h&72h
(续表)
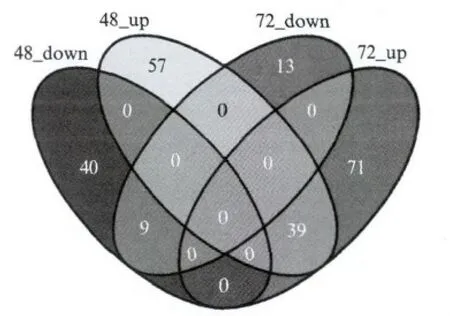
图2 Ang Ⅱ处理48h和72h时上调和下调的基因数目Fig.2 The number of genes both up-regulation induced by AngⅡfor 48hand 72h
为了进一步研究受到AngⅡ显著调控的基因在48 h和72h两种处理中上调和下调的情况,我们将差异表达基因的标准变得更为严格(变化幅度超过2倍并且P≤0.01).分析发现,有39个基因在48h和72h中均被上调;57个基因只在48h处理时上调,不受72h处理影响;71个基因只在72h处理时上调,不受48h处理的影响;仅仅有9个基因在48h和72h处理时均被下调;40个基因只在48h处理时下调,不受72h处理影响;13个基因只在72h 处理时上调,不受48h 处理的影响(图2).这些结果显示在48h和72h共同上调的基因数目比下调的基因数目多,而48h或72h特异上调的基因数目也比对应时间特异下调的基因数目多.
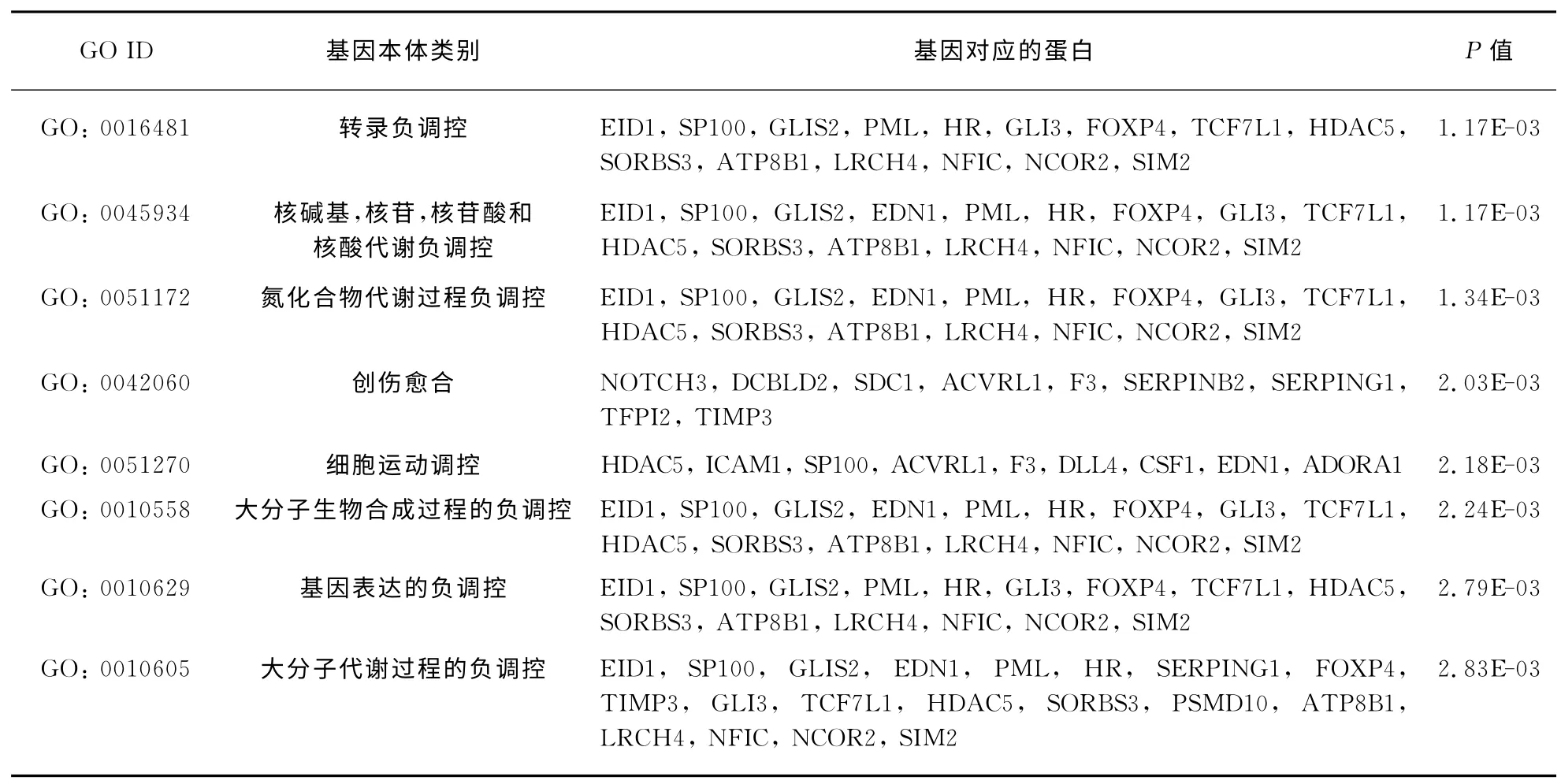
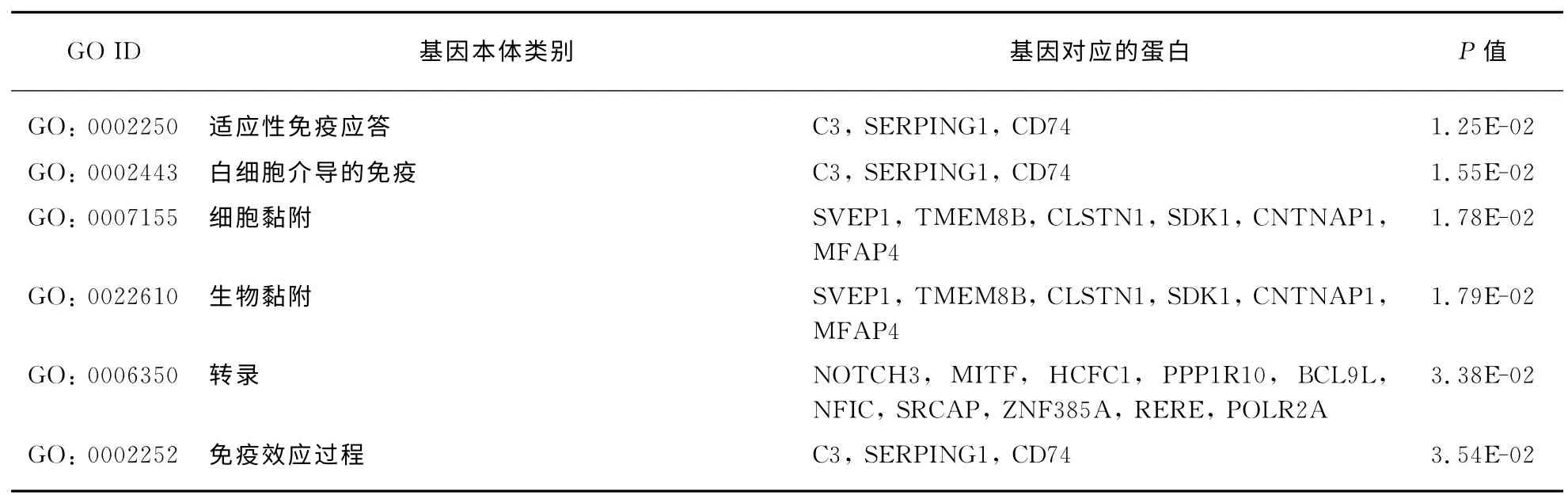
对于在AngⅡ_48h和AngⅡ_72h处理时均上调的39个基因进行GO 生物学过程富集分析(表3)发现,在富集的前十个GO 生物学功能中有3个与细胞黏附、转录相关,这都是VSMC衰老过程中的细胞和分子层面发生的相关现象.其余7个GO 生物学功能都是与免疫调控相关的,这提示细胞在受到外界刺激时,会启动一系列免疫调控过程,保护细胞使之免受这些刺激的破坏,维持健康的状态.这也从另一个角度说明Ang Ⅱ诱导hVSMC的处理是成功的.

表3 AngⅡ处理48h&72h均上调的基因GO 生物学功能富集分析Tab.3 GO-analysis of genes both up-regulation induced by Ang Ⅱfor 48h&72h
(续表)
通过对衰老的hVSMC中显著差异表达的基因进行分子功能(Molecular Function,MF)分析,我们发现在AngⅡ_48h处理中富集的前5个都是与酶活性相关的(表4).而AngⅡ_72h中差异表达的基因都富集在和金属离子相关的功能中,例如钠离子,锌离子及其调控它们的蛋白(表5).Ang Ⅱ刺激的Ca2+信号很复杂,通过多条信号途径整合而成.一方面,细胞内的Ca2+促进Ca2+信号的快速短暂释放[20].另一方面,细胞外Ca2+引起的Ca2+的持续升高与Ang Ⅱ刺激的持续血管收缩有关[21,22].此外,Ang Ⅱ刺激细胞内Na+的升高,Mg+的降低以及Na+/H+交换[23].

表4 AngⅡ处理48h后的GO 分子功能富集分析Tab.4 GO-MF analysis of genes inAng Ⅱ_48hinduction

表5 AngⅡ处理72h后的GO 分子功能富集分析Tab.5 GO-MF analysis of genes in Ang Ⅱ_72hinduction
2.3 TGF-β信号通路在AngⅡ诱导的人平滑肌细胞衰老中受损
本研究对于48h和72h处理后差异表达的基因也进行了信号通路的分析,但并没有发现显著富集的信号通路.一种可能的推测是信号通路主要在蛋白质层面而非RNA 层面参与人平滑肌细胞的诱导衰老.另一种可能是只有通路中的少数基因发生了RNA 水平的改变,因此相应的通路并没有达到显著水平.于是我们把目光转向与细胞衰老有关的各种信号通路,通过对选定的信号通路进行系统分析来判断该通路是否参与了平滑肌细胞的衰老.
衰老的细胞会发展出一系列复杂的促炎症反应,即衰老相关的分泌表型(Senescence-Associated Secretory Phenotype,SASP)[24-27].转化生长因子-β(TGF-β)是SASP中重要的部分,通过SMAD 复合体上调细胞周期抑制子p27和p15的表达[28].TGF-β可以通过旁分泌的方式引起邻近细胞发生衰老[29,30].研究表明,Ang Ⅱ在细胞内产生的ROS是许多TGF-β调控反应的重要传递分子[31,32],然而活性氧物质会选择性降低TβRⅡ和Smad3的蛋白水平[33],进而损伤TGF-β信号通路.Ang Ⅱ处理48h时TGF-β1的表达量略有下降,72h的时候TGF-β1的表达回升到基底水平(图3(a)).RNA-seq的数据显示TGF-β的跨膜受体TβRⅡ和细胞内受体TβRI都有所下降(图3(a)).此外,RNA-seq的结果显示TGF-β信号通路的靶基因CCN2(别名CTGF)在Ang Ⅱ诱导时降低,qPCR 结果也表明CCN2在Ang Ⅱ处理48h和72h时RNA 水平均降低了55%左右(图3(b)),这与文献报道一致[33].
RNA-seq数据显示p27和p15,两个仅仅受TGF-β/SMAD3调控的细胞周期抑制子,它们的表达都明显降低(图3(c)(d)).研究采用实时定量PCR(qPCR)对这两个靶基因进行了验证,发现p27的RNA水平在48h和72hAngⅡ处理中都下调了56%左右,p15的RNA 水平在48h处理时下调15%,在72h处理时下调了28%(图3(c)(d)).这些结果提示在Ang Ⅱ处理诱导的hVSMC 中,TGF-β信号通路有可能发生了损伤,从而使得下游靶基因表达量下降.
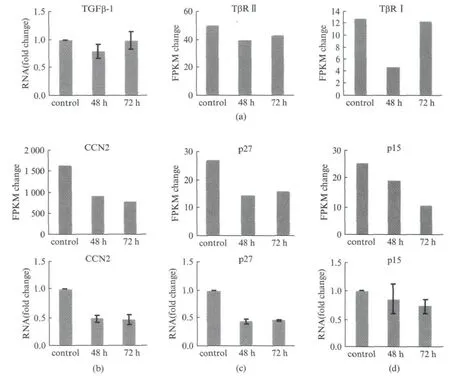
图3 Ang Ⅱ处理hVSMC中TGF-β信号通路受损Fig.3 TGF-βsignal pathway is impaired in AngⅡ-induced hVSMC
2.4 p38-p53-p21通路参与调控hVSMC的衰老
许多文献报道了在其他细胞系里,p38MAPK 蛋白激酶的磷酸化水平是Ang Ⅱ激活信号通路的一个关键的部分[34],为了验证在hVSMC 里的这一关键分子是否也起作用,我们检测了该激酶的磷酸化水平变化.Western blot的结果显示,在Ang Ⅱ处理48h和72h时,p38磷酸化水平明显升高(图4(a)),而且,处理时间越长,升高越明显.而RNA-seq的结果显示p38编码基因的RNA 水平在48h基本没变化,72h时略有下降(图4(a)).p38作为蛋白激酶,一方面可以活化p53蛋白,p53进一步磷酸化激活p21蛋白,另一方面又调控p16 的活性[28].此外,p38 激酶MKK3(MAPKK3)的蛋白质水平也在Ang Ⅱ处理中升高,而其编码基因的RNA 水平在48h略有上升,72h时又回降到基底水平(图4(b)).
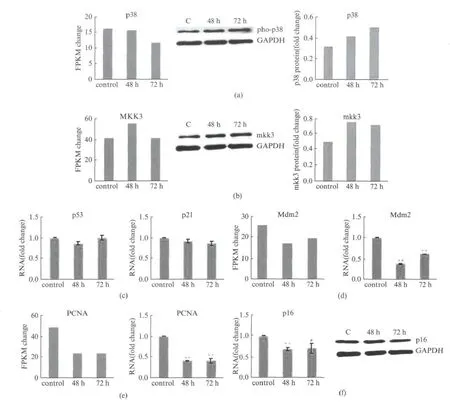
图4 p38信号通路参与Ang Ⅱ诱导的hVSMC衰老Fig.4 p38signal pathway involves in AngⅡ-induced hVSMC
然而有意思的是,在衰老的hVSMC中,p53除了磷酸化水平受p38上调外,p53的稳态蛋白质水平也明显升高(图1(b)).稳态蛋白质水平明显升高的还有另一个衰老的重要标记物p21(图1(b)),但p21和p53的RNA 水平却都没有太大变化(图4(c)).这一结果暗示在Ang Ⅱ诱导的hVSMC 中,衰老标志物p53、p21是在蛋白质水平或蛋白质修饰水平而非RNA 水平进行调控.为了进一步明确其潜在的调控机制,我们研究了特异调控p53蛋白水平的基因.Mdm2(mouse double minute 2homolog),是泛素化降解p53蛋白的一个非常重要的酶蛋白.RNA-seq结果和qPCR 验证均显示,在AngⅡ处理的hVSMC中,Mdm2编码基因的RNA 水平在48h时降低63%,72h时有所回升,但与对照相比,仍然降低了40%(图4(d)).增殖细胞核抗原(Proliferating Cell Nuclear Antigen,PCNA)是DNA 滑动钳家族的成员,有持续的高合成能力,能够将DNA 聚合酶δ连接到DNA 模板上,这对于整个基因组的复制非常重要.研究发现,在Ang Ⅱ诱导的hVSMC中,PCNA 编码基因RNA 水平降低了60%(图4(e)).而p38-p53-p21信号通路正是PCNA 编码基因转录调控的上游,推测由于该信号通路的激活,使得CyclinE-CDK2介导的RB蛋白去磷酸化,PCNA 编码基因启动子区域进一步结合抑制复合物,PCNA 基因表达水平降低,DNA 无法完成复制,导致细胞周期停滞,进而引起了hVSMC的衰老.
值得注意的是,qPCR 的结果显示在AngⅡ诱导的hVSMC中,p16的RNA 水平在48h和72h时都降低了30%(图4(f)),Western blot的结果也证实了p16蛋白在48h和72hAngⅡ处理中是降低的,说明Ang Ⅱ刺激并不是通过p16通路诱导hVSMC衰老的.以上的证据支持p38-p53-p21信号通路参与了Ang Ⅱ诱导的人血管平滑肌细胞的衰老,而与之平行的p38-p16信号通路则并不参与该衰老调控过程.
3 讨论
在100nmol/L Ang Ⅱ处理的hVSMC 48h和72h中,细胞衰老的标志物β-半乳糖苷酶染色都呈现蓝色,而且在72h的时候染色的细胞增多更明显.衰老的标志物p53和p21蛋白在48h和72h处理时表达都明显升高,随着处理时间的延长,升高更明显.此外,我们进一步发现PCNA 在Ang Ⅱ处理48h和72h时都明显下调.PCNA 通过将DNA 聚合酶δ连接到DNA 模板上,完成DNA 的复制,使得细胞周期可以顺利进行,完成细胞分裂.PCNA 的下调导致了基因组DNA 无法顺利完成复制,细胞周期发生停滞.在这种损伤无法得到修正的情况下,细胞发生衰老.
不少文献报道在细胞衰老过程中,TGF-β1信号通路被激活[35].TGF-β1是免疫调节细胞因子,调控免疫细胞的增殖、存活、分化和迁移.在免疫系统中,TGF-β1通过调控淋巴细胞增殖、分化和存活来诱导和增强免疫耐受力的提高.TGF-β1的损坏引起免疫系统发生紊乱[36],从而导致不正常的免疫细胞活化、炎症反应、癌症(白血病,恶性肿瘤等)、以及自身免疫疾病[37].事实上,TGF-β1缺陷小鼠由于淋巴细胞和单核细胞侵入多个重要器官,在出生2~3周后就会死亡[38].然而在本研究中,TGF-β1的靶基因CCN2的表达在Ang Ⅱ48h和72h处理时都被下调,仅仅只受TGF-β信号通路调控的p27和p15在Ang Ⅱ处理48h和72h时均明显下调,这些结果提示在AngⅡ处理诱导的hVSMC中,TGF-β信号通路有可能发生了损伤,这与文献报道一致[33],从而使得下游靶基因表达量下降.文献报道激活的TGF-β信号通路可通过上调p21,p27和p15的表达量来抑制Rb蛋白磷酸化并进而促进细胞衰老[28].而p27和p15的表达量在Ang Ⅱ处理后下调,因此推测受损的TGF-β信号通路可能不参与Ang Ⅱ诱导的hVSMC 细胞衰老.
在Ang Ⅱ诱导的hVSMC中,MKK3激酶和p38蛋白激酶表达升高,而MKK6并没有表达,说明在hVSMC中Ang Ⅱ刺激p38的磷酸化水平升高是由MKK3介导的,从而开启了p38信号通路的活化.活化的p38磷酸化p53蛋白,后者又进一步上调p21.值得一提的是,Mdm2基因RNA 水平和蛋白质水平在48h和72hAng Ⅱ处理时均明显下降.一方面,Mdm2泛素化p53,使之运出细胞核外[39],调控p53的蛋白活性;另一方面,过表达的Mdm2能够促进p53依赖蛋白酶体的降解[40].由此我们可以推断,在AngⅡ诱导的hVSMC衰老中,p53主要是通过稳态蛋白质水平和磷酸化水平而非RNA 水平进行调控的.
伴随着个体衰老,血管紧张素-Ⅱ信号级联反应在动脉中逐渐放大[15],刺激VSMC 产生炎症因子和促血栓因子,从收缩型转变成分泌型,向细胞外基质分泌大量的蛋白,引起VSMC 发生重塑,并进而促进衰老.衰老的VSMC发生迁移,导致血管变厚,僵硬度增加,从而增加多种衰老相关的疾病的发生率,例如高血压、动脉粥样硬化、中风等.总的来说,本研究较为详细地研究了Ang Ⅱ诱导的人血管平滑肌细胞衰老中关键因子的表达情况,发现TGF-β信号通路发生了受损,细胞周期抑制子p16同样发生了下调,而p38-p53-p21参与了细胞的衰老调控,为理解人血管平滑肌细胞诱导性衰老的调控机制提供了重要参考.
[1]LAKATTA E G,LEVY D.Arterial and cardiac aging:Major shareholders in cardiovascular disease enterprises:Part I:aging arteries:a“set up”for vascular disease[J].Circulation,2003,107(1):139-146.
[2]MARIN J.Age-related changes in vascular responses:A review[J].Mech Ageing Dev,1995,79(2-3):71-114.
[3]BRANDES R P,FLEMING I,BUSSE R.Endothelial aging[J].Cardiovasc Res,2005,66(2):286-294.
[4]NAJJAR S S,SCUTERI A,LAKATTA E G.Arterial aging:Is it an immutable cardiovascular risk factor?[J].Hypertension,2005,46(3):454-462.
[5]NAKAJIMA M,HASHIMOTO M,WANG F,et al.Aging decreases the production of PGI2in rat aortic endothelial cells[J].Exp Gerontol,1997,32(6):685-693.
[6]COMI P,CHIARAMONTE R,MAIER J A.Senescence-dependent regulation of type 1plasminogen activator inhibitor in human vascular endothelial cells[J].Exp Cell Res,1995,219(1):304-308.
[7]HOFFMANN J,HAENDELER J,AICHER A,et al.Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli:important role of nitric oxide[J].Circ Res,2001,89(8):709-715.
[8]MATSUSHITA H,CHANG E,GLASSFORD A J,et al.eNOS activity is reduced in senescent human endothelial cells:Preservation by hTERT immortalization[J].Circ Res,2001,89(9):793-798.
[9]VAUX D L,STRASSER A.The molecular biology of apoptosis[J].Proc Natl Acad Sci USA,1996,93(6):2239-2244.
[10]HERBERT K E,MISTRY Y,HASTINGS R,et al.Angiotensin Ⅱ-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways[J].Circ Res,2008,102(2):201-208.
[11]KUNIEDA T,MINAMINO T,NISHI J,et al.AngiotensinⅡinduces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway[J].Circulation,2006,114(9):953-960.
[12]MIN L J,MOGI M,IWANAMI J,et al.Cross-talk between aldosterone and angiotensin Ⅱin vascular smooth muscle cell senescence[J].Cardiovasc Res,2007,76(3):506-516.
[13]de CAVANAGH E M,PIOTRKOWSKI B,FRAGA C G.Concerted action of the renin-angiotensin system,mitochondria,and antioxidant defenses in aging[J].Mol Aspects Med,2004,25(1/2):27-36.
[14]BASSO N,PAGLIA N,STELLA I,et al.Protective effect of the inhibition of the renin-angiotensin system on aging[J].Regul Pept,2005,128(3):247-252.
[15]NAJJAR S S,SCUTERI A,LAKATTA E G.Arterial aging:is it an immutable cardiovascular risk factor?[J].Hypertension,2005,46(3):454-462.
[16]KIM D,PERTEA G,TRAPNELL C,et al.TopHat2:accurate alignment of transcriptomes in the presence of insertions,deletions and gene fusions[J].Genome Biol,2013,14(4):R36.
[17]LANGMEAD B.Aligning short sequencing reads with Bowtie[J].Curr Protoc Bioinformatics,2010,11:11-17.
[18]POLLIER J,ROMBAUTS S,GOOSSENS A.Analysis of RNA-Seq data with TopHat and Cufflinks for genome-wide expression analysis of jasmonate-treated plants and plant cultures[J].Methods Mol Biol,2013,1011:305-315.
[19]DIMRI G P,LEE X,BASILE G,et al.A biomarker that identifies senescent human cells in culture and in aging skin in vivo[J].Proc Natl Acad Sci USA,1995,92(20):9363-9367.
[20]TOUYZ R M,SCHIFFRIN E L.Role of calcium influx and intracellular calcium stores in angiotensinⅡ-mediated calcium hyper-responsiveness in smooth muscle from spontaneously hypertensive rats[J].J Hypertens,1997,15(12Pt 1):1431-1439.
[21]RUAN X,ARENDSHORST W J.Calcium entry and mobilization signaling pathways in ANG Ⅱ-induced renal vasoconstriction in vivo[J].Am J Physiol,1996,270(3Pt 2):F398-F405.
[22]INSCHO E W,IMIG J D,COOK A K.Afferent and efferent arteriolar vasoconstriction to angiotensinⅡand norepinephrine involves release of Ca2+from intracellular stores[J].Hypertension,1997,29(1Pt 2):222-227.
[23]TOUYZ R M,SCHIFFRIN E L.Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensinⅡin vascular smooth muscle cells[J].Pharmacol Rev,2000,52(4):639-672.
[24]EVAN G I,D’ADDA D F F.Cellular senescence:Hot or what?[J].Curr Opin Genet Dev,2009,19(1):25-31.
[25]CAMPISI J.Aging,cellular senescence,and cancer[J].Annu Rev Physiol,2013,75:685-705.
[26]COPPE J P,DESPREZ P Y,KRTOLICA A,et al.The senescence-associated secretory phenotype:The dark side of tumor suppression[J].Annu Rev Pathol,2010,5:99-118.
[27]KUILMAN T,PEEPER D S.Senescence-messaging secretome:SMS-ing cellular stress[J].Nat Rev Cancer,2009,9(2):81-94.
[28]MUNOZ-ESPIN D,SERRANO M.Cellular senescence:From physiology to pathology[J].Nat Rev Mol Cell Biol,2014,15(7):482-496.
[29]ACOSTA J C,BANITO A,WUESTEFELD T,et al.A complex secretory program orchestrated by the inflammasome controls paracrine senescence[J].Nat Cell Biol,2013,15(8):978-990.
[30]NELSON G,WORDSWORTH J,WANG C,et al.A senescent cell bystander effect:Senescenceinduced senescence[J].Aging Cell,2012,11(2):345-349.
[31]JIANG Z,SEO J Y,HA H,et al.Reactive oxygen species mediate TGF-beta1-induced plasminogen activator inhibitor-1upregulation in mesangial cells[J].Biochem Biophys Res Commun,2003,309(4):961-966.
[32]RHYU D Y,YANG Y,HA H,et al.Role of reactive oxygen species in TGF-beta1-induced mitogenactivated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells[J].J Am Soc Nephrol,2005,16(3):667-675.
[33]HE T,QUAN T,SHAO Y,et al.Oxidative exposure impairs TGF-beta pathway via reduction of typeⅡreceptor and SMAD3in human skin fibroblasts[J].Age(Dordr),2014,36(3):9623.
[34]USHIO-FUKAI M,ALEXANDER R W,AKERS M,et al.p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin Ⅱ.Role in vascularsmooth muscle cell hypertrophy[J].J Biol Chem,1998,273(24):15022-15029.
[35]MORALES M G,VAZQUEZ Y,ACUNA M J,et al.AngiotensinⅡ-induced pro-fibrotic effects require p38MAPK activity and transforming growth factor beta 1expression in skeletal muscle cells[J].Int J Biochem Cell Biol,2012,44(11):1993-2002.
[36]MCKARNS S C,SCHWARTZ R H.Distinct effects of TGF-beta 1on CD4+and CD8+T cell survival,division,and IL-2 production:A role for T cell intrinsic Smad3[J].J Immunol,2005,174(4):2071-2083.
[37]SHULL M M,ORMSBY I,KIER A B,et al.Targeted disruption of the mouse transforming growth factor-beta 1gene results in multifocal inflammatory disease[J].Nature,1992,359(6397):693-699.
[38]KULKARNI A B,HUH C G,BECKER D,et al.Transforming growth factor beta 1null mutation in mice causes excessive inflammatory response and early death[J].Proc Natl Acad Sci USA,1993,90(2):770-774.
[39]LOHRUM M A,WOODS D B,LUDWIG R L,et al.C-terminal ubiquitination of p53contributes to nuclear export[J].Mol Cell Biol,2001,21(24):8521-8532.
[40]HAUPT Y,MAYA R,KAZAZ A,et al.Mdm2promotes the rapid degradation of p53[J].Nature,1997,387(6630):296-299.
