聚乙二醇-聚唾液酸嵌段聚合修饰尿酸酶
2015-11-11卢绍曾吴剑荣郑志永詹晓北
卢绍曾,吴剑荣,朱 莉,郑志永,詹晓北
(江南大学 生物工程学院 糖化学与生物技术教育部重点实验室,江苏 无锡 214122)
聚乙二醇-聚唾液酸嵌段聚合修饰尿酸酶
卢绍曾,吴剑荣,朱 莉,郑志永,詹晓北
(江南大学 生物工程学院 糖化学与生物技术教育部重点实验室,江苏 无锡 214122)
探索一种综合聚唾液酸(PSA)和聚乙二醇(PEG)优势的蛋白修饰方法。对纯化的聚唾液酸进行两步活化,先在非还原端氧化产生活性醛基,再加入胱胺形成活性巯基;活化的聚唾液酸(相对分子质量为3.4×104)和异基双功能的PEG(相对分子质量为3.5×103)形成嵌段聚合物,然后于4℃修饰尿酸酶。利用凝胶层析(Toyopearl HW-55F)对修饰后的尿酸酶进行纯化,收集相应峰进行化学法和SDS-PAGE电泳鉴定,经多角度激光光散射凝胶系统测定缀合物相对分子质量为5.214×105;相对于原始酶,修饰酶酶活保留率72.4%,体外热失活半衰期由115.5 h提高到231 h,对高温、酸碱、胰蛋白酶的耐受稳定性显著提高。
聚乙二醇;聚唾液酸;嵌段聚合;尿酸酶;缓释
目前,随着生物医药技术的发展,各种蛋白及多肽类药物更好地保障了人类健康。2012年,该类药物总销售额达1 246亿美元,且每年都继续稳定增长。但是,蛋白类药物存在着稳定性差、易被酶降解、半衰期短、生物利用度低等缺点,非人源蛋白自身具有免疫原性,进入人体易引起抗体反应,从而加速血液清除,降低了药效[1]。对蛋白质类药物的改造成为当下研究的热点,目前比较成熟、成功的方法是聚乙二醇(PEG)修饰。PEG修饰使用较多的是单甲氧基聚乙二醇(mPEG)修饰剂,目前已经应用在包括尿酸酶[2-3]、天冬酰胺酶[4]、胰岛素[5]、白细胞介素-2[6]、CC49/218单链抗体[7]、干扰素α-2b[8]和肿瘤坏死因子纳米抗体[9]等蛋白及多肽上。修饰后蛋白稳定性普遍提高,半衰期延长,免疫原性降低,生物利用度提高。由于PEG为人工合成,相对分子质量较大,其很难通过肾小球被尿液排出,在体内降解非常缓慢,造成聚乙二醇在机体内的积累[10]。已有研究证明,聚乙二醇在溶酶体内的积累具有毒性[11],且慢性治疗中,频繁注射聚乙二醇化蛋白易产生抗体[12]。
与此同时,采用聚唾液酸(PSA)作为蛋白修饰剂的研究也在进行。聚唾液酸是由唾液酸单体(N-乙酰神经氨酸)以α-2,8糖苷键连接成的聚阴离子多糖[13],其单体广泛存在于人和动物体内细胞表面的寡糖链末端,因而表现出良好的生物相容性、生物可降解性及抗人体免疫系统识别功能[14]。目前,常见的方法是还原氨基化,已在研究的蛋白包括过氧化氢酶[15]、天冬酰胺酶[16-17]、胰岛素[18]、超氧化物歧化酶[19]及丁酰胆碱酯酶[20]等。聚唾液酸化蛋白不存在免疫原性,但修饰程度往往不及聚乙二醇化的修饰程度,且聚唾液酸水化能力稍弱。
尿酸酶能将尿酸降解为溶解度高其13倍的尿囊素,在治疗顽固性痛风及高尿酸血症方面有较好的效果。但作为外源蛋白,尿酸酶在体内易被酶水解,稳定性低、免疫原性较强,因而限制了它的应用。本研究中笔者结合PEG与PSA的优点,以尿酸酶为对象,采用全新的嵌段聚合修饰方法,将活化的聚唾液酸与小分子PEG形成嵌段聚合物,用于修饰尿酸酶,使其兼具聚唾液酸的抗免疫识别和PEG的强吸水性能[21],以提高稳定性和半衰期。
1 材料与方法
1.1 材料
聚唾液酸为笔者所在实验室成员发酵纯化所得(E.coli K235),数均相对分子质量3.42×104;尿酸酶,来源于假丝酵母基因,在大肠杆菌中表达,SDS-PAGE鉴定相对分子质量为3.5×104。尿酸、5,5′-二硫双(2-硝基苯甲酸)(DTNB)购自南京都莱有限公司;异基双功能PEG(MAL-PEG-NHS)购自北京键凯科技有限公司,相对分子质量为3 592;蛋白Marker、胰蛋白酶(1∶300)购自上海生工工程有限公司;透析袋(7000)、超滤离心管(3000)购自上海基星科技有限公司;其他试剂均购自国药集团化学试剂公司,化学纯。
1.2 方法
1.2.1 聚唾液酸的活化
称取纯化的PSA 200mg溶于50mL加盖试管,加入10mL 0.1 mol/L高碘酸钠,室温避光振荡15min,加10mL乙二醇终止反应,继续振荡30min,然后在0.02%(质量分数)(NH4)2CO3透析液中4℃透析24 h。透析液用超滤离心管浓缩,加入100倍摩尔比的胱胺盐酸盐,滴加0.1 mol/L NaOH调pH 7.4,37℃磁力搅拌1 d,加入与胱胺盐酸盐等摩尔的二硫苏糖醇,37℃反应1 h,反应液用相对分子质量为3 000的离心超滤管4 000g×30min超滤8次,除去未反应的小分子,浓缩得到非还原端官能团为巯基的活性唾液酸[21]。
1.2.2 聚唾液酸活化程度测定
5,5′-二硫代双(2-硝基苯甲酸)(DTNB)法:将DTNB溶于50 mmol/L pH 7.0的Na2HPO4中制成10 mmol/L的DTNB标准液。将标准液用0.25 mol/L pH 8.3的Tris-HCl缓冲液稀释100倍制得DTNB分析液。将活化的PSA稀释至1mL,分别加入到5mL预先恒温于25℃水浴中的DTNB分析液,摇匀,静置10min,立即于波长412 nm处测定吸光值(A),以半胱氨酸为标准。通过DTNB法测定巯基含量(mmol)后,再除以PSA的量(mmol)即可获得聚唾液酸的活化程度。
1.2.3 PEG-PSA嵌段聚合修饰尿酸酶
按照摩尔比1∶1,将具有活性巯基的PSA与PEG在25℃反应0.5 h,然后按摩尔比50∶1加入尿酸酶,于4℃冰水浴反应2 h。
1.2.4 修饰尿酸酶的纯化
采用分子筛柱进行纯化。柱子尺寸:16mm× 270mm;填料:TOSOH Toyopearl HW-55F;流动相:20 mmol/L pH 7.4 H3PO4缓冲液,0.15 mol/L NaCl;洗脱条件:流速0.7mL/min;检测器:示差折光检测器串联紫外检测器(λ=215 nm)。
1.3 缀合物的表征
1.3.1 温度稳定性
将等量天然酶和修饰酶置于50和60℃水浴保温,定时(1、2、3、4、5和6 h),取100 μL置于冰浴测定酶活,以各温度下0 h的原始酶活为对照。
1.3.2 酸碱稳定性
取等量天然酶和修饰酶,在37℃,pH为6.0、7.0(H3PO4缓冲液)、8.0、9.0和10.0(H3BO3缓冲液)水浴2 h后测定酶活(25℃,pH 8.5 H3BO3缓冲液)。以各pH下0 h的原始酶活为对照。
1.3.3 抗酶解稳定性
取500 μL 0.1mg/mL尿酸酶,按体积比10∶1加入胰蛋白酶液(1%,20 mmol/L、pH 6.8 H3PO4缓冲液),在37℃水浴保持0、0.5、1、2和3 h后取样测定酶活,计算酶活保留率。
1.3.4 酶动力学参数测定
取180 μL不同浓度尿酸溶液(40~140 μmol/L)于石英96孔板,加入0.1mg/mL尿酸酶在25℃反应3min,每15 s记录1次293 nm波长处的吸光值,计算反应速率。最大反应速率和米氏常数的测定采用Lineweave-Burk双倒数作图。
1.4 尿酸酶酶活的测定
根据尿酸于293 nm处有特征吸收,测定尿酸被尿酸酶氧化生成尿囊素后,剩余尿酸在293 nm处的吸光值。酶活测定反应体系均为0.1 mol/L H3BO3缓冲液(pH 8.5),以0.001%的尿酸为底物,25℃反应3min,每15 s记录一次293 nm波长处的吸光值。以每分钟催化1 μmol尿酸分解所需要的酶量定义为1个酶活力单位(U)。
1.5 聚唾液酸含量测定
聚唾液酸含量采用间苯二酚法,以N-乙酰神经氨酸为标品[22]。
1.6 蛋白含量测定
蛋白质含量测定参照文献[23]进行,以牛血清白蛋白为参照。
1.7 聚乙二醇含量测定
将待测溶液稀释至4mL,然后加入1mL 5% BaCl2和0.5mL 0.1 mol/L碘溶液,15min后在535 nm处测定吸光值[24],以 PEG(相对分子质量4 000)为参照(0~15mg/L)。
1.8 相对分子质量测定
1.8.1 聚唾液酸相对分子质量测定
聚唾液酸相对分子质量采用十八角度激光光散射凝胶系统(Wyatt Technology,USA)进行监测。色谱柱为Shodex-Ohpak SB-804 HQ串联Ohpak SB-802 HQ,流动相为0.1 mol/L NaNO3,流速为0.5mL/min,检测器为Wyatt DAWN HELEOSⅡ十八角度激光光散射仪串联Optilab T-rEX示差折射率检测器,标品为T40,相对分子质量4.0×104。
1.8.2 尿酸酶、缀合物相对分子质量测定
聚唾液酸-聚乙二醇修饰尿酸酶缀合物的相对分子质量采用十八角度激光光散射凝胶系统进行监测,色谱柱为Shodex-Ohpak SB-806 HQ,流动相为0.1 mol/L NaNO3,流速为0.4mL/min,检测器为Waters UV/VISIBLE串联Wyatt DAWN HELEOSⅡ十八角度激光光散射仪串联Optilab T-rEX示差折射率检测器。
1.9 SDS-PAGE电泳分析
SDS-PAGE电泳用于监测尿酸酶修饰前后的相对分子质量变化,采用质量分数12%分离胶和5%浓缩胶[25]。
2 结果与讨论
2.1 聚唾液酸活化
聚唾液酸非还原端C7~9位有3个相邻的醇羟基,反应活性较弱,通过活化得到活性巯基,进而与PEG修饰剂的马来酰亚胺官能团发生加成反应,形成嵌段聚合物。先用高碘酸钠特异性地氧化醇羟基得到醛基,接着与胱胺盐酸盐反应,最后用二硫苏糖醇还原,切断二硫键,得到末端巯基。经过透析和离心超滤两步纯化,累积回收率为(68.2± 4.5)%(n=3),活化程度为(38.6±2.68)%(n=3)。由于聚唾液酸是大分子,本身活化不易进行,且产生的末端巯基在空气中不是很稳定,较易被氧化,因此活化程度不是特别高。
2.2 PEG-PSA嵌段聚合修饰尿酸酶
活化的聚唾液酸先与双官能团PEG修饰剂的马来酰亚胺发生加成反应,形成嵌段聚合物,然后PEG的另一官能团(NHS)与尿酸酶上赖氨酸残基的氨基发生亲电取代反应,形成酰胺键。尿酸酶通过嵌段聚合修饰后,相对分子质量增大,未修饰酶及其他副产物相对分子质量则较小,反应产物通过分子筛层析进行纯化,结果如图1所示。由图1可知:按照示差信号共收集4个峰,对收集的峰进行化学鉴定,分别测定其聚唾液酸、聚乙二醇和蛋白含量。
发现只有第1个峰检测到聚唾液酸,其他3个峰未检出;第1和第2个峰均检测到聚乙二醇,第3和第4个峰未检出,且峰1的聚唾液酸含量最高;第1、第2和第4个峰均检测到蛋白,且峰1蛋白含量最高。TOSOH Toyopearl HW-55F分子筛色谱柱填料是由甲基丙酸烯聚合而成的基质,主要依靠分子流体力学半径分离物质,嵌段聚合修饰尿酸酶后,尿酸酶的水化半径显著增大,所以第1个出峰。第2个流出峰为少量聚乙二醇连接的蛋白,第3个流出峰为溶剂峰,第4个峰只检测到蛋白,且215 nm处有紫外吸收,应为没有修饰的尿酸酶,可能与填料上的羧基存在静电相互作用,所以最后出峰。

图1 分子筛层析纯化尿酸酶修饰产物结果Fig.1 Gel permeation chromatography of modified uricase
2.3 缀合物的表征结果分析
2.3.1 SDS-PAGE结果分析
除了进行化学鉴定,还对收集的峰进行进一步SDS-PAGE分析,结果如图2所示。由图2可知:未修饰尿酸酶单体相对分子质量约3.5×104,纯化收集第1个峰为修饰产物,单体相对分子质量约4.5×104,还有一模糊条带为5.5×104,可能还有更高相对分子质量的产物(由于比例较低,未出现条带)。多条带的出现可能是由于嵌段聚合修饰是随机修饰,造成产物的不均一性。由相对分子质量半对数—迁移率曲线所计算得到的相对分子质量并不能反映修饰产物的真实相对分子质量,因为PEG吸水较强,导致泳动速率较慢,得到的相对分子质量偏大[3];但同时作为嵌段聚合物之一的PSA是阴离子多糖,与十二烷基磺酸(SDS)所带电荷一致,导致修饰产物可能泳动速度较快[26]。总之,修饰产物相对分子质量的电泳行为同时受到PEG和PSA的影响。第2个峰由于蛋白含量较低,未出现条带。第4个峰的相对分子质量与单体相对分子质量一致,分析为未被修饰的尿酸酶。
2.3.2 PSA-PEG修饰尿酸酶的相对分子质量表征及生化参数
通过多角度激光光散射凝胶系统测定修饰尿酸酶的相对分子质量,结果如图3所示。

图2 纯化产物电泳分析Fig.2 SDS-PAGE analysis of the purified uricase from GPC

图3 尿酸酶-PEG-PSA缀合物的相对分子质量分布Fig.3 Molecular weight distribution of modified uricase-PEG-PSA conjugate
一般尿酸酶是四聚体(相对分子质量1.412× 105),由图3可知:在连接上PSA-PEG后,相对分子质量变成5.214×105。PSA和PEG的相对分子质量为3.42×104和3.5×103,通过计算(5.214×105-1.412×105)/(3.42×104+3.5×103)=10.08,则平均每个尿酸酶单体连接2.52个嵌段聚合物修饰剂。尽管尿酸酶每个单体含有32个赖氨酸残基,但是理论上只有10~12是可以接近的。可能是这些赖氨酸残基相距较近,嵌段聚合物连在其中一个位点,由于空间位阻,其他修饰剂将无法连接[27-28]。此外,聚唾液酸是阴离子多糖,同时还存在电荷排斥,因此,修饰程度可能不及PEG修饰。蛋白质化学修饰往往是随机修饰,可能与活性部位的赖氨酸发生作用,导致酶活降低,经测定,嵌段聚合修饰后酶活保留率为72.4%。Zhang等[29]采用含羟基琥珀酰亚胺官能团的mPEG 350和mPEG 5k,修饰来自苛求芽胞杆菌的尿酸酶,酶活保留率分别为15%和65%;Bomalaski等[30]用PEG 5000 MW修饰假丝酵母尿酸酶,酶活保留率50%。在37℃恒温孵育箱测定体外半衰期,酶的热失活半衰期由115.5 h提高到231 h,提高1倍。Zhang等[29]采用PEG修饰后,在40℃热失活半衰期由40 h提高至85 h,嵌段聚合修饰结果与其接近。
2.3.3 PSA-PEG修饰尿酸酶的温度稳定性
大部分蛋白酶在较高温度下,天然构象遭到破坏,从紧密有序趋向随即松散,折叠结构打开,导致酶活性丧失。尿酸酶-PEG-PSA缀合物的温度稳定性如图4所示。

图4 尿酸酶-PEG-PSA缀合物的温度稳定性Fig.4 Stability of uricase-PEG-PSA conjugate under different temperature
由图4可知:在50℃下,2 h内修饰酶基本未损失酶活,原始酶则损失了15%的酶活,6 h后修饰酶酶活保留率为72%,而原始酶则仅为60%。更高的温度(60℃)下,修饰酶和原始酶酶活损失都较大,但修饰酶在6 h后酶活剩余34%,原始酶则为27%。整体而言,修饰后尿酸酶比原始尿酸酶对温度的耐受性更强,这有利于酶的储运,即使短暂暴露于较高的温度条件下,亦能保留较多酶活。推测原因,可能是酶的活性位点受到嵌段聚合物的保护,提高了尿酸酶蛋白的刚性[31]。
2.3.4 PSA-PEG修饰尿酸酶的酸碱稳定性
pH对酶稳定性具有重要的影响,反应体系pH的变化,会影响酶活性部位的基团解离状态,从而影响酶的活性。极端的pH会使维护酶三维结构的部分——非共价键受到干扰,导致酶自身的变性。考察尿酸酶-PEG-PSA缀合物的酸碱稳定性,结果见图5。
由图5可知:在不同pH条件下处理2 h后,修饰尿酸酶酶活保留率高于原始酶,其中在碱性条件下较为明显。在pH 10时,修饰酶比原始酶酶活高出22%,而在接近人体血液pH条件下,修饰酶残余酶活70%,原始酶活则为60%,修饰酶更加稳定,显示出良好的临床应用前景。在酸性条件下,修饰酶和原始酶酶活损失都较多,因为尿酸酶属过氧化物酶,在碱性pH条件下有最大酶活,酸性条件下酶易降解失活。另外,修饰剂之一的聚唾液酸在碱性条件下稳定,酸性条件下糖苷键也会被水解[14],所以酸性条件下酶的稳定性提高不多。

图5 尿酸酶-PEG-PSA缀合物的酸碱稳定性Fig.5 pH stability of uricase-PEG-PSA conjugate
2.3.5 PSA-PEG修饰尿酸酶的酶解稳定性
蛋白酶药物进入体内后,由于体内较多的蛋白酶存在,使得酶蛋白被水解,多肽链断裂,蛋白酶的功能结构受到破坏而丧失活性,因此必须考察尿酸酶修饰后对蛋白酶的稳定性,结果如图6所示。
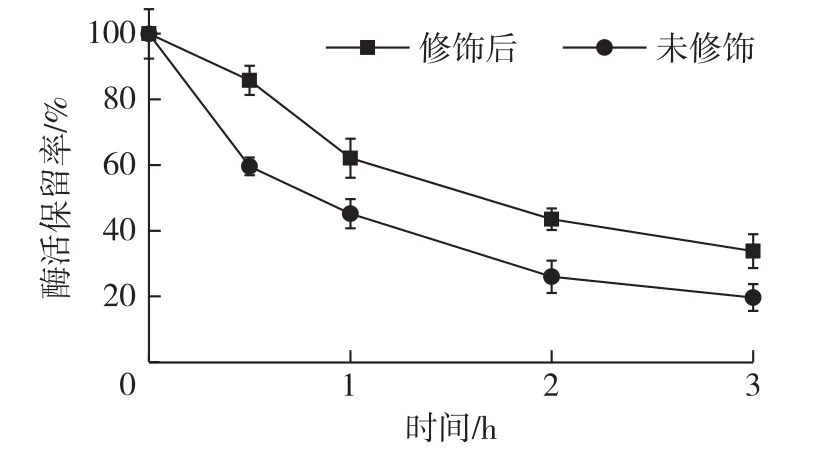
图6 尿酸酶-PEG-PSA缀合物的抗酶解稳定性Fig.6 Protease resistant stability of uricase-PEG-PSA conjugate
由图6可知:在0.5 h时,修饰尿酸酶酶活残余85%,而原始酶为60%,3 h后修饰尿酸酶残余酶活34%,原始酶则只剩20%。尿酸酶修饰后,对胰蛋白酶的耐受性显著增加,稳定性提高,在体内能更加持久地发挥药效,可减少给药剂量,降低治疗成本,同时也可减少病人的痛苦(目前临床尿酸酶的给药途径主要是静脉注射)。推测原因,可能是尿酸酶上暴露在外面的蛋白酶识别位点被嵌段聚合修饰剂共价连接后,识别位点被掩蔽,无法被蛋白酶识别,进而降解,另外聚唾液酸具有较强的负离子性,与酶蛋白周围带相反电荷的氨基酸残基相互作用,形成第二层保护层[19]。
2.3.6 PSA-PEG修饰尿酸酶的动力学参数
分别比较原始尿酸酶与修饰尿酸酶的动力学参数,结果如表1所示。由表1可以看出,尿酸酶修饰后,米氏常数Km减小,对底物的亲和能力增强,最大反应速率Vmax增大,催化效率提高。Zhang等[29]用mPEG 350和mPEG 5k修饰尿酸酶后Km没有明显的变化。尿酸酶修饰后并没有因为嵌段聚合修饰剂的屏蔽效应,造成底物与酶亲和能力的下降,推测原因,可能是酶的溶解度较低[32],但是PEG的强吸水性能加上聚唾液酸的高度亲水性,增加了酶的溶解度,使酶与底物的亲和能力增强。酶修饰后最大反应速率与催化效率均有所增加。

表1 尿酸酶的动力学参数Table 1 Kinetic parameters of modified uricase
3 结论
本研究确定了一种以聚唾液酸-聚乙二醇嵌段聚合物修饰蛋白的方法。采用异基双功能聚乙二醇-MAL-PEG-NHS为接头,将聚唾液酸与尿酸酶链接起来,平均每个尿酸酶单体交联2.52个PEG-PSA嵌段聚合物,酶活保留率为72.4%,热失活半衰期由115.5 h提高到231 h。另外,米氏常数Km由原始酶的239.7 μmol/L降为162.6 μmol/L,最大反应速率Vmax由原始酶的83.3 μmol/(L·min)增大为90.9 μmol/(L·min),kcat由1.94 s-1增大为2.12 s-1。PSAPEG嵌段聚合修饰蛋白程度不如PEG修饰,但由于其易降解,不产生免疫反应,可用于进一步临床实验,在未来可能是一种替代PEG修饰的更好方法。
[1]Pisal D S,Kosloski M P,Balu-Iyer S V.Delivery of therapeutic proteins[J].J Pharm Sci,2010,99(6):2557-2575.
[2]Schlesinger N,Yasothan U,Kirkpatrick P.Pegloticase[J].Nature Rev Drug Discov,2010,10(1):17-18.
[3]Zhang C,Fan K,Luo H,et al.Characterization,efficacy,pharmacokinetics,and biodistribution of 5kDa mPEG modified tetrameric canine uricase variant[J].Int J Pharm,2012,430(1):307-317.
[4]Veronese F M,Monfardini C,Caliceti P,et al.Improvement of pharmacokinetic,immunological and stability properties of asparaginase by conjugation to linear and branched monomethoxy poly(ethylene glycol)[J].J Contr Release,1996,40(3):199-209.
[5]Jain S,Hreczuk-Hirst D H,McCormack B,et al.Polysialylated insulin:synthesis,characterization and biological activity in vivo[J].Biochim Biophys Acta,2003,1622(1):42-49.
[6]Goodson R J,Katre N V.Site-directed pegylation of recombinant interleukin-2 at its glycosylation site[J].Nature Biotechnol,1990,8(4):343-346.
[7]Lee L S,Conover C,Shi C,et al.Prolonged circulating lives of single-chain Fv proteins conjugated with polyethylene glycol:a comparison of conjugation chemistries and compounds[J]. Bioconjug Chem,1999,10(6):973-981.
[8]Ramon J,Saez V,Baez R,et al.PEGylated interferon-α2b:a branched 40K polyethylene glycol derivative[J].Pharm Res,2005,22(8):1375-1387.
[9]Vugmeyster Y,Entrican C A,Joyce A P,et al.Pharmacokinetic,biodistribution,and biophysicalprofilesofTNF nanobodies conjugated to linear or branched poly(ethylene glycol)[J]. Bioconjug Chem,2012,23(7):1452-1462.
[10]Roberts M J,Bentley M D,Harris J M.Chemistry for peptide and protein PEGylation[J].Adv Drug Deliv Rev,2012,54:459-476.
[11]Gregoriadis G,Fernandes A,Mital M,et al.Polysialic acids:potential in improving the stability and pharmacokinetics of proteins and other therapeutics[J].Cell Mol Life Sci,2000,57(13/14):1964-1969.
[12]Ŝroda K,Rydlewski J,Lnagner M,et al.Repeated injections of PEG-PE liposomes generate anti-PEG antibodies[J].Cell Mol Biol Lett,2005,10:37-47.
[13]郑志永,詹晓北,吴剑荣,等.大肠杆菌产生的聚唾液酸的结构[J].食品与生物技术学报,2004,24(5):38-41.
[14]McGuire E J,Binkley S B.The structure and chemistry of colominic acid[J].Biochemistry,1964,3(2):247-251.
[15]Fernandes A I,Gregoriadis G.Synthesis,characterization and properties of sialylated catalase[J].Biochim Biophys Acta,1996,1293(1):90-96.
[16]Fernandes A I,GregoriadisG.Polysialylated asparaginase:preparation,activity and pharmacokinetics[J].Biochim Biophys Acta,1997,1341(1):26-34.
[17]Fernandes A I,Gregoriadis G.The effect of polysialylation on the immunogenicity and antigenicity of asparaginase:implication in its pharmacokinetics[J].Int J Pharm,2001,217(1):215-224.
[18]Jain S,Hreczuk-Hirst D H,McCormack B,et al.Polysialylated insulin:synthesis,characterization and biological activity in vivo[J].Biochim Biophys Acta,2003,1622(1):42-49.
[19]Wu J R,Lin Y,Zheng Z Y,et al.Improvement of the CuZnsuperoxide dismutase enzyme activity and stability as a therapeutic agent by modification with polysialic acids[J]. Biotechnol Lett,2010,32(12):1939-1945.
[20]Ilyushin D G,Smirnov I V,Belogurov A A,et al.Chemical polysialylation of human recombinant butyrylcholinesterase delivers a long-acting bioscavenger for nerve agents in vivo[J]. Proc Nat Acad Sci USA,2013,110(4):1243-1248.
[21]吴剑荣,詹晓北,林怡,等.聚乙二醇-聚唾液酸嵌段共聚物的制备方法及应用:中国:101780117 A[P].2010-07-28.
[22]Svennerholm L.Quantitative estimation of sialic acids[J]. Biochem Biophys Acta,1957,24(4):604-611.
[23]Brown R E,Jarvis K L,Hyland K J.Protein measurement using bicinchoninic acid:elimination of interfering substances[J].Anal Biochem,1989,180(1):136-139.
[24]Skoog B.Determination of polyethylene glycols 4000 and 6000 in plasma protein preparations[J].Vox Sanguinis,1979,37(6):345-349.
[25]Laemmli U K.Cleavage of structural proteins during the assembly of the head of bacteriophage T4[J].Nature,1970,227:680-685.
[26]Constantinou A,Epenetos A A,Hreczuk-Hirst D,et al.Modulation of antibody pharmacokinetics by chemical polysialylation[J]. Bioconjug Chem,2008,19(3):643-650.
[27]Caliceti P,Schiavon O,Veronese F M.Immunological properties of uricase conjugated to neutral soluble polymers[J].Bioconjug Chem,2001,12(4):515-522.
[28]Sherman M R,Saifer M G P,Perez-Ruiz F.PEG-uricase in the management of treatment-resistant gout and hyperuricemia[J]. Adv Drug Deliv Rev,2008,60(1):59-68.
[29]Zhang C,Yang X,Feng J,et al.Effects of modification of amino groups with poly(ethylene glycol)on a recombinant uricase from Bacillus fastidiosus[J].Biosci Biotechnol Biochem,2010,74(6):1298-1301.
[30]Bomalaski J S,Holtsberg F W,Ensor C M,et al.Uricase formulated with polyethylene glycol(Uricase-PEG 20):biochemical rationale and preclinical studies[J].Rheumatol,2002,29(9):1942-1949.
[31]Soares A L,Guimarães G M,Polakiewicz B,et al.Effects of polyethylene glycol attachment on physicochemical and biological stability of E.coli L-asparaginase[J].Int J Pharm,2002,237(1):163-170.
[32]Kelly S J,Delnomdedieu M,Oliverio M I,et al.Diabetes insipidus in uricase-deficient mice:a model for evaluating therapy with poly(ethylene glycol)-modified uricase[J].J Am Soc Nephrol,2001,12(5):1001-1009.
(责任编辑 管 珺)
Uricase modification with polysialic acid and polyethylene glycol(PEG-PSA)co-polymer
LU Shaozeng,WU Jianrong,ZHU Li,ZHENG Zhiyong,ZHAN Xiaobei
(Key Labroratory of Carbohydrate Chemistry&Biotechnology of the Ministry of Education,School of Biotechnology,Jiangnan University,Wuxi 214122,China)
A novel protein modification method was developed by integrating the advantages of polysialic acid(PSA)and polyethylene glycol(PEG).PSA was activated by two steps:the aldehyde group was generated by periodate oxidation at the non-reducing end,and then the active thiol group was formed using cystamine.The activated PSA(3.4×104)reacted with hetero-bifunctional PEG(3.5×103)to form a block co-polymer,and modified uricase at 4℃.The conjugate was purified with gel permeation chromatography and the target peak was collected and identified with chemical method and SDS-PAGE. The molecular weight of the conjugate was 5.214×105through multi-angle laser light scattering gel system.Compared with native enzyme,residual enzyme activity was 72.4%,and heat inactivation half life in vitro was improved from 115.5 h up to 231 h.The tolerance stability from heat,acid and alkaline,trypsin treatment was significantly improved.
polyethyleneglycol;polysialic acid;block polymerization;uricase;controlled-release
Q55
A
1672-3678(2015)02-0086-07
10.3969/j.issn.1672-3678.2015.02.017
2014-01-25
江苏省自然科学基金(BK2011158)
卢绍曾(1987—),男,河南郑州人,硕士研究生,研究方向:发酵工程;吴剑荣(联系人),副教授,E-mail:kinowu76@163.com
