人参固本丸的质量标准研究
2015-10-27北京同仁堂股份有限公司科学研究所100079彭鹏
北京同仁堂股份有限公司科学研究所(100079)彭鹏
人参固本丸[1]收载于《中华人民共和国卫生部药品标准》第一册,由人参、地黄、熟地黄、山茱萸(酒炙)、山药、泽泻、牡丹皮、茯苓、麦冬、天冬等十味药组成。该方主要功效是滋阴益气,固本培元,用于阴虚气弱,虚劳咳嗽,心悸气短,骨蒸潮热,腰酸耳鸣,遗精盗汗,大便干燥。原标准只收载了茯苓、山药、人参、熟地黄、山茱萸、泽泻、牡丹皮的显微鉴别,不足以对本方进行质量控制,本研究增加了人参[2]、山茱萸[2]、麦冬[2]的薄层鉴别,同时采用高效液相色谱法测定山茱萸中马钱苷的含量。通过研究,该方法简便易行,准确可靠。
1 仪器与材料
①仪器:岛津LC-10AVP高效液相色谱仪。色谱柱:ZORBAX SB-C18 (5μm 150mm×4.6mm)。②材料:人参固本丸样品由北京同仁堂天然药物有限公司提供,批号:3012999、4012002、5012995、5012996。马钱苷(批号为:111640-200503)、人参皂苷Rg1(110703-200425)、人参皂苷Re(110754-200421)、熊果酸(批号742-200212)、丹皮酚(110708-200506),麦冬(批号:121013-200506),均购自中国药品生物制品检定所。实验所用试剂均为分析纯,液相色谱所用试剂均为色谱纯。
2 方法与结果
2.1 薄层鉴别

附图1 人参的薄层色谱图
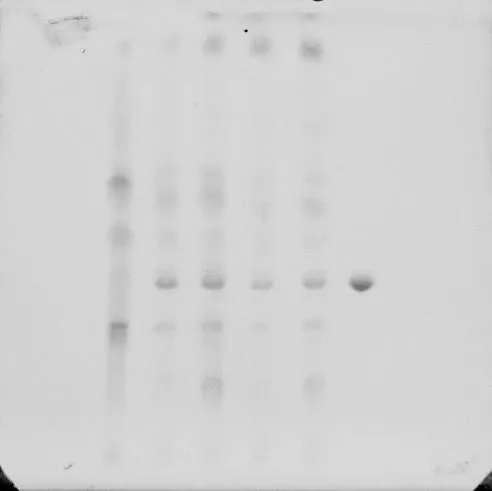
附图2 山茱萸的薄层色谱图

附图3 麦冬的薄层色谱图
2.1.1 人参的薄层鉴别 取本品15g,剪碎,加硅藻土5g,研匀,加乙醚40ml,超声处理10分钟,滤过,弃去乙醚液,药渣挥干乙醚,加水饱和的正丁醇50ml,超声处理30分钟,滤过,滤液加1%氢氧化钠溶液洗涤两次,每次15ml,弃去碱液,再加正丁醇饱和的水洗至中性,正丁醇液蒸干,残渣加甲醇0.5ml使溶解,作为供试品溶液。另取人参皂苷Rg1对照品、人参皂苷Re对照品,加甲醇制成每1ml各含1mg的混合溶液;照薄层色谱法(《中国药典》2010年版一部附录ⅥB)试验,吸取上述供试品溶液6μl、对照品溶液2μl,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(13∶7∶2)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。结果见附图1。
2.1.2 山茱萸的薄层鉴别 取本品20g,剪碎,加硅藻土10g,研匀,加乙醚60ml,超声处理15分钟,滤过,滤液挥干乙醚,残渣加丙酮1ml使溶解,作为供试品溶液。另取熊果酸对照品,加乙酸乙酯制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2010年版一部附录ⅥB)试验,吸取对照品溶液及供试品溶液各6μl,分别点于同一硅胶G薄层板上,以甲苯-乙酸乙酯-甲酸(20∶4∶0.5)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。
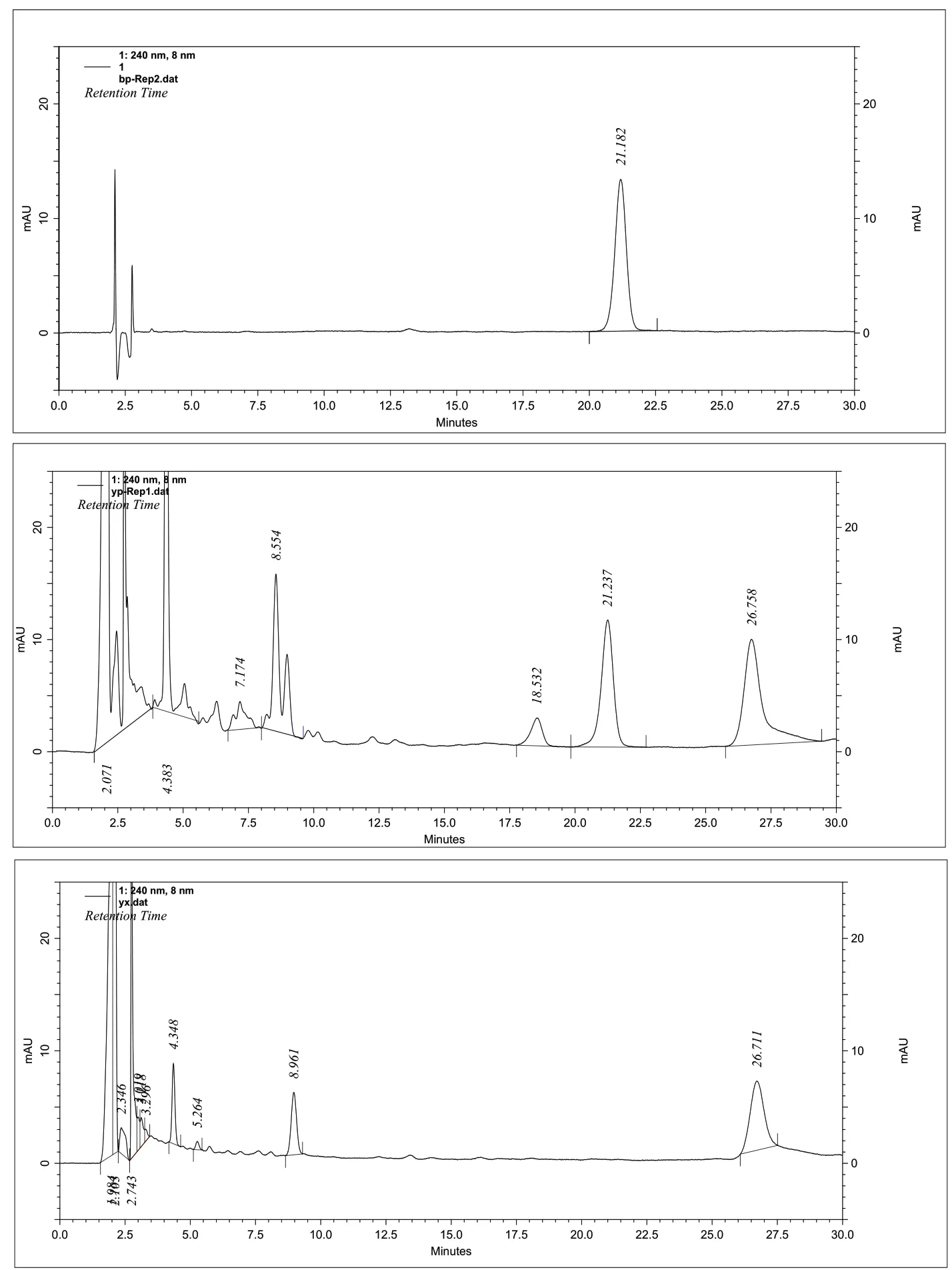
附图4 人参固本丸中马钱苷高效液相色谱图
2.1.3 麦冬的薄层鉴别 取本品10g ,剪碎,加水饱和的正丁醇40ml,超声处理20分钟,滤过,滤液蒸干,残渣加水30ml溶解,再加盐酸3ml,置沸水浴上加热回流1小时,放冷,用三氯甲烷提取2次,每次20ml,合并三氯甲烷液,蒸干,残渣加乙醇1ml使溶解,作为供试品溶液。另取麦冬对照药材1g,同法制成对照药材溶液。照薄层色谱法(《中国药典》2005年版一部附录ⅥB)试验,吸取上述两种溶液5μl,分别点于同一硅胶G薄层板上,以三氯甲烷-丙酮(4∶1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。
2.2 含量测定 ①色谱条件 色谱柱:ZORBAX SB-C18(5μm 150mm×4.6mm)色谱柱;流动相:乙腈-甲醇-0.1%磷酸溶液(8∶4∶88);检测波长:240nm;柱温:40℃;流速:0.8ml/min;理论板数按马钱苷峰计算应不低于5000。②对照品溶液的制备 取马钱苷对照品适量,精密称定,加50%甲醇制成每1mL含20μg的溶液,即得。③供试品溶液的制备 取重量差异检查项下的本品,剪碎,取约2g,精密称定,置具塞锥形瓶中,精密加入50%甲醇25ml,密塞,称定重量,超声处理(功率250W,频率33kHz)15分钟使溶散,加热回流1小时,放冷,再称定重量,用50%甲醇补足减失的重量,摇匀,滤过。精密量取续滤液10ml,置中性氧化铝柱(100目~200目,4g,内径1cm,干法装柱)上,用40%甲醇50ml洗脱,收集流出液及洗脱液,蒸干,残渣加50%甲醇适量使溶解,并转移至10ml量瓶中,加50%甲醇稀释至刻度,摇匀,即得。④空白试验 按处方中药味比例,自配不含山茱萸的群药,按标准中制法制成空白制剂,照供试品溶液制备方法制样,测定,结果阴性对照样品在与马钱苷相同保留时间处,未见色谱峰,故认为无干扰。
⑤线性关系考察 精密吸取浓度为0.02452mg/ml的马钱苷对照品溶液3、6、9、12、15、20ul,及浓度为0.1226 mg/ml的马钱苷对照品溶液6、8、10、12 ul注入液相色谱仪,照上述色谱条件测定,以峰面积积分值为纵坐标,马钱苷进样量为横坐标,绘制标准曲线,得回归方程:y= 2057008.79 x + 12650.04 r=0.9999结果表明,马钱苷在0.07356~1.4712μg范围内具良好的线性关系。⑥精密度试验 取同一供试品溶液(5012996批)连续进样6次,观察仪器的精密度,测得马钱苷含量的平均值为0.2189mg/g,相对标准偏差RSD=0.45%。⑦稳定性试验 取同一供试品溶液(5012996批),分别于配制后0、2、4、8、12、24小时,依法测定,马钱苷平均总量为0.2156mg/g,相对标准偏差RSD=1.30%(n=6),试验结果表明,供试品溶液制备后24小时内基本稳定。⑧重复性试验 取同批(5012996)样品,平行制样6份,测定,求得马钱苷平均含量为0.2112mg/g,相对标准偏差分别为RSD=0.89%(n=6)。⑨回收率试验 采用加样回收法,精密称取已知含量的同批(5012996,马钱苷含量0.2112mg/g)样品1g,分别精密加入马钱苷对照品(0.28198mg),照2.2.3项下方法操作,平行制样6份,测定,按下式计算回收率,结果表明本方法具有良好的准确度。测得马钱苷的平均回收率为101.56%,相对标准偏差RSD=1.31%(n=6)。⑩马钱苷对照品纯度测定 精密称取马钱苷对照品10mg,置50mL棕色量瓶中,加50%甲醇溶解并稀释至刻度,摇匀,吸取对照品溶液5μL注入液相色谱仪中,归一化法测定含量,马钱苷含量为100%。11样品测定结果 分别取4批样品,依上述色谱条件进行测定,每批样品测定2次,测定结果见附表2。
3 讨论
本次研究增加了人参、山茱萸、麦冬的薄层鉴别,通过对多批样品的鉴别,结果表明薄层色谱鉴别方法良好,阴性无干扰,可以作为人参固本丸的质量控制方法之一。
含量测定的供试品溶液制备中,根据马钱苷的性质,参考《中国药典》2010版一部山茱萸项下含量测定项,在30、60、90分钟不同回流时间进行了比较,结果表明提取60分钟后马钱苷的含量不再增加,因此确定提取时间为60分钟。比较了加入提取溶剂的量对马钱苷含量的影响,通过比较,在加入10ml、25ml、50ml时,用25ml与50ml溶剂提取的马钱苷含量相近,故选择25ml溶剂进行提取。
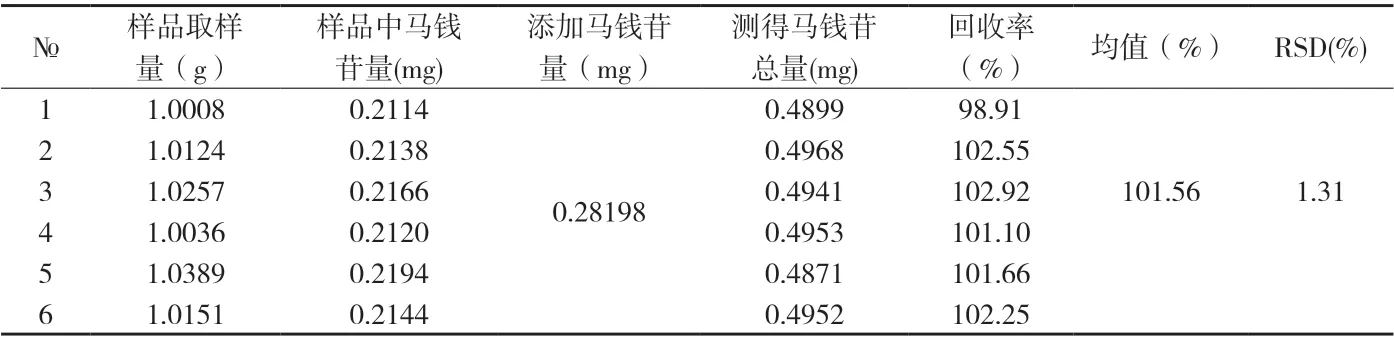
附表1 马钱苷回收率试验成果

附表2 样品中马钱苷测定结果(n=2)
由于处理方法中有过中性氧化铝柱的步骤,为保证洗脱完全,在50ml洗脱液流尽后再加入10ml洗脱液,收集后测定含量,通过测定,后续的10ml样品液中马钱苷的含量为0,结果表明50ml洗脱液已将样品完全洗净。
检测波长的选择:取马钱苷对照品,加50%甲醇溶解,在200~400nm范围内扫描,结果在240nm处有最大吸收,故选择240nm作为检测波长。
通过本次研究,完善了人参固本丸的质量标准,增加了薄层鉴别及含量测定项,与原来仅有显微鉴别的标准相比,提高了质量控制水平,有利于保证药品的质量。
