敏捷食酸菌腈水解酶基因在大肠杆菌中的表达
2015-10-25管启旺马江锋贺爱永姜岷宫长斌
管启旺 马江锋 贺爱永 姜岷 宫长斌
(南京工业大学生物与制药工程学院 材料与化学工程国家重点实验室,南京 211816)
敏捷食酸菌腈水解酶基因在大肠杆菌中的表达
管启旺 马江锋 贺爱永 姜岷 宫长斌
(南京工业大学生物与制药工程学院 材料与化学工程国家重点实验室,南京 211816)
以含有敏捷食酸菌的腈水解酶基因的克隆质粒为模板,通过PCR扩增获得长度为1 080 bp的腈水解酶(Nitrilase)基因片段。使用表达质粒pET-28-a构建表达载体,获得重组质粒pET-Nit。对所表达的基因进行测序对比发现与GenBank所公布的基因序列比较,相似性99%,读码框出现2 bp的突变,但并未引起相应氨基酸突变,故不影响酶的表达与特性。将重组质粒转化到表达宿主Escherichia coli Rosetta(DE3)感受态中,使用诱导剂IPTG对菌株进行诱导表达,获取菌液进行SDS-PAGE分析,得知目的蛋白分子量约为41.28 kD,与预期所想的一致。酶活力分析表明,上清的比活力为3 U/mg。进一步对诱导条件进行优化,包括诱导温度,IPTG浓度,诱导时间等一系列条件。在最优条件下,扩大培养体积,比活力可达15 U/mg,活力提高了5倍左右。
腈水解酶;敏捷食酸菌;大肠杆菌;克隆
腈水解酶(EC 3.5.5.1,Nitrilase)是一类催化腈类物质水解的特异性酶,属于腈水解酶超家族[1]。该酶兼具有腈水合酶(EC 4.2.1.84,nitrile hydratase)催化腈类水解成酰胺,以及酰胺水解酶(EC 3.5.1.4,amidase)催化酰胺水解成羧酸和氨两种功能,它能直接催化腈类物质水解成相应的羧酸并释放出氨气[2]。
腈水解酶的水解反应机制(图1),酶促反应的中心是一个半胱氨酸残基,半胱氨酸巯基的强亲核性使得整个酶促反应类似于碱条件下的酸碱催化,整个反应包含了两分子的水的结合与一分子氨气的脱离[3]。腈水解酶的活性不受大多数金属离子和螯合剂的影响,但Ag+和Hg+等重金属离子能和巯基作用,对酶的活性有较大影响,这证明了半胱氨酸的巯基在反应中的重要作用。Brenner等[4]根据蠕虫的NitFhit的晶体结构分析,认为腈水解酶一族的催化活性中性,是一个含谷氨酸-赖氨酸-半胱氨酸(Glu-Lys-Cys)的脆性催化三联体(catalytic triad)。
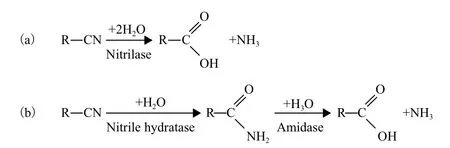
图1 腈水解酶(a)与腈水合酶酰胺水解酶(b)催化底物水解机制
由于腈水解酶具有高度的区域以及立体选择性,使得该酶在化工和药物合成等领域越来越受关注[5]。腈水解酶的作用底物主要分为以下几类:脂肪和芳香族脂肪腈,芳香腈,脂环和杂环腈,二腈等腈类化合物。例如,研究脂肪和芳香族脂肪腈水解,用于乙醇酸的生产[6,7];以芳腈作为底物时,萘普生、布洛芬和氟比洛芬等α-芳基丙酸化合物以及R-扁桃酸的生产[8];脂环和杂环类的腈转化,比较有特色的是利用红球菌属实现烟酸的生产以及有关拟南芥的腈水解酶用于合成吲哚-3-乙酸的研究[9];二腈的选择性水解比较代表性的是,普瑞巴林合成过程中手性中间体的生产[10]。
目前该酶的研究主要集中在国外,国内有关研究还处于较落后阶段,有关双腈的选择性催化水解的文章基本较少,亟待筛选高产该类酶的菌株或通过基因手段构建相关的工程菌[11],Acidovorax facilis ATCC 55746是目前报道的选择性水解双腈活力突出的菌株[12]。本研究选用在大肠杆菌中有高表现的pET系统,构建pET-Nit重组质粒,实现成功诱导表达,并对发酵产酶的诱导相关条件进行系列优化,优化后酶产量提高了5倍,能达到产酶的先进水平。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒 表达质粒pET-28a和表达宿主E.coli Rosetta(DE3)由本实验室自主保藏;敏捷食酸菌腈水解酶基因片段由上海生物工程公司合成(基因克隆在PUC57质粒上,插入位点为BamH I)。
1.1.2 主要试剂 异丙基硫代-β-D-半乳糖苷(IPTG)购自BioDev公司;限制性内切酶Nde I,Sal I,DL15000 DNA Maker,Solution I高效连接酶等购自TaKaRa公司;小量质粒提取和胶回收试剂盒购自AXYGEN公司;PCR引物由金斯瑞合成;其余试剂为国产或者进口分析纯。
1.1.3 培养基 LB培养基(g/L):胰蛋白胨10,酵母粉5,氯化钠10(固体平板加入琼脂粉20)。SOC培养基(g/L):20胰蛋白胨,5酵母粉,0.5氯化钠,0.2 氯化钾,0.94氯化镁,1.6葡萄糖。
1.2 方法
1.2.1 引物设计和腈水解酶的克隆 根据敏捷食酸菌ATCC 55746腈水解酶基因序列(GenBank 登录号为DQ444267.1),设计上下游扩增引物:P1:5'-GCTCATATGGTTTCGTATAACAGCAAGTTCCTCG -3';P2:5'-GGTGTCGACCTTTGCTGGGACCGGTTCTT-3'。在上下游引物分别引入Nde I和Sal I酶切位点(下划线部分),将原始基因的起始密码子GTG改为大肠优选的ATG,去除末端的终止密码子使得表达蛋白带上纯化标签,并同时在引物前方加入3个bp的保护碱基。预计扩增产物长度约为1 080 bp的基因片段。引物由金斯瑞(南京)公司合成。
提取含有敏捷食酸菌腈水解酶基因的质粒作为扩增模板,利用上述引物进行PCR扩增。PCR反应条件为:盖温99℃,94℃预变性5 min,94℃变性45 s,56℃退火45 s,72℃延伸2 min,工作时间30个循环。
1.2.2 重组表达质粒的构建和测序对比 PCR产物与质粒pET-28a使用Nde I/Sal I内切酶双酶切过夜,胶回收酶切产物。将纯化的目的基因片段和线性化载体按适当比例,用Solution I高效连接酶连接过夜,使用氯化钙转化法进行感受态转化。筛选阳性重组子,培养和提取质粒进行PCR验证与测序。经鉴定正确的重组质粒命名为pET-Nit。通过Blast对测序结果进行对比分析。
1.2.3 腈水解酶酶液的提取 挑取重组大肠杆菌E.coli Rosetta(DE3)/pET-Nit单菌落,接种到含有50 mg/L硫酸卡那霉素的5 mL的 LB液体培养基中,37℃,250 r/min过夜培养后,按1%接种量接种到新鲜的含有50 mg/L硫酸卡那霉素的50 mL 的LB液体培养基中,37℃培养3 h后,加入IPTG进行诱导表达。
菌液冷冻离心(4℃,8 000 r/min,20 min),弃去上清,沉淀使用磷酸钾缓冲液清洗(pH7.0,100 mmol/L的浓度)两次后,超声破碎(功率300 W,超声3 s,间隔5 s,工作时间3 min),超声后菌液冷冻离心(4℃,8 000 r/min,20 min)。弃去沉淀取上清液测定酶活力。
1.2.4 SDS-PAGE分析 取方法1.2.3中的酶液进行SDS-PAGE分析。采用12.5%的分离胶和4%的浓缩胶制胶,电泳后使用考马斯亮蓝R-250进行染色。以原始宿主菌和未经诱导的重组菌株的破碎上清作为对照。
1.2.5 腈水解酶活力测定 酶活力测定使用苯酚硫酸盐显色法,显色液与标准液的制备方法如文献[13]所述。取酶液20 μL,加入到10 mL含有100 mmol/L最适底物反丁烯二腈的磷酸钾缓冲液(pH7.0,100 mmol/L)中(反丁烯二腈使用N,N-二甲基甲酰胺当助溶剂,终浓度为5%),酶促反应 30 min后取出使用硫酸铵作为标准样,测定铵根离子的浓度。一个酶活力单位(U)定义为:每分钟能催化1 μmol底物水解所需的酶量。
蛋白质含量测定使用Brandford法[14],以牛血清白蛋白为标准样。腈水解酶比活力定义为:每毫克蛋白所具有的酶活单位数。
2 结果
2.1 Nit基因的克隆与分析
扩增腈水解酶基因,PCR产物经琼脂糖凝胶电泳检测(图2),在1 080 bp左右具有很清晰的条带,与预期设计一致。提取质粒对目的基因测序分析,目的片段与GenBank所报道的基因相似性为99%,在酶读码框的第996和1 032位的碱基G和A发生突变分别突变为T和G,对应的332和343位的氨基酸仍然是缬氨酸和谷氨酸,并不影响酶的表达和性质。
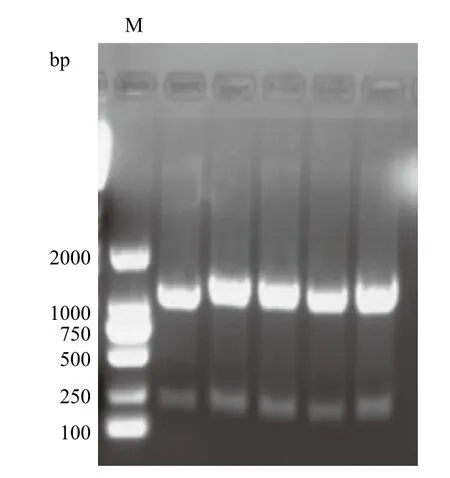
图2 目的片段PCR扩增
2.2 重组质粒的构建和表达
构建的表达质粒经Nde I/Sal I双酶切鉴定(图3),pET-Nit在1 080 bp左右比空载体明显多出一个条带,与插入目的基因大小相同,表明片段已经成功插入,重组质粒构建成功。

图3 pET-Nit重组质粒双酶切验证
2.3 腈水解酶的诱导表达和SDS-PAGE分析
重组菌pET-Nit/Rosetta(DE3)经过0.2 mmol/L的IPTG在30℃诱导表达12 h后,腈水解酶比酶活为3 U/mg,而空宿主不具备酶活,未诱导的重组菌表达量几乎为零。SDS-PAGE电泳结果显示(图4),重组菌株经诱导表达后在41.28 kD处有明显的融合条带,与预期的判断一致,出发菌和未诱导的在该区域不具有明显条带。根据酶活测定和SDS-GAGE结果显示,腈水解酶Nit已经在重组大肠杆菌中成功表达。
2.4 诱导条件优化
通过对诱导温度,诱导物浓度和诱导时间进行优化,找到了工程菌的最佳产酶条件。
2.4.1 诱导温度优化 加入IPTG后分别在20、25、30、35和40℃下进行诱导,时间为12 h,结果显示(图5)低温时候比酶活更高,这表明低温有助于酶的正确折叠,不易形成包涵体,但是温度过低,对菌体生长速度有限制[15]。
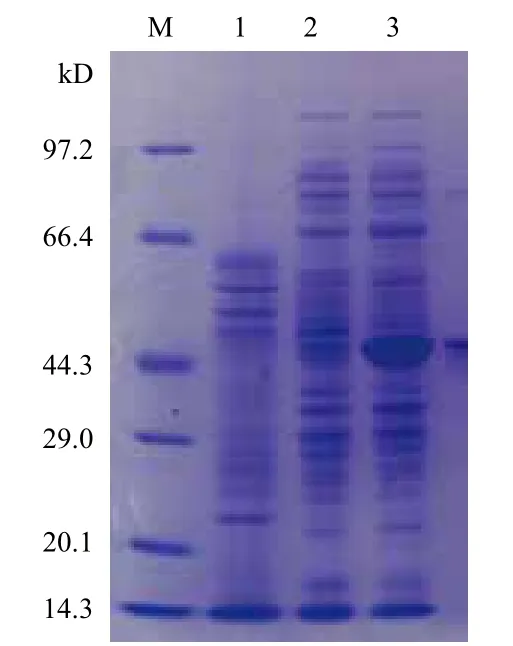
图4 pET-Nit SDS-PAGE 电泳图谱(全细胞蛋白)
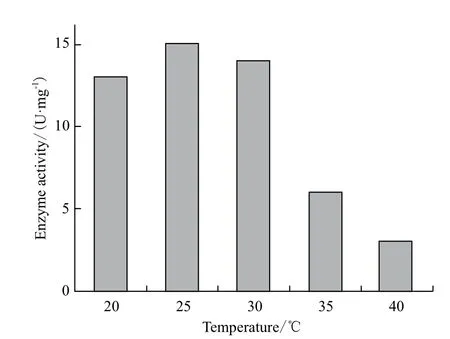
图5 诱导温度优化
2.4.2 IPTG浓度的优化 分别加入0.05、0.1、0.2、0.4、0.8及1.2 mmol/L终浓度的IPTG,25℃诱导表达12 h。结果(图6)显示,加入量为0.2 mmol/L IPTG时菌体密度最佳,并且比酶活最高,为15 U/mg。IPTG浓度过小,表达量和菌体浓度都不够,而IPTG浓度过大菌体量和酶活力反而下降,这表明不能为菌体代谢的高浓度的IPTG会抑制菌体生长。2.4.3 IPTG诱导时间优化 加入IPTG至终浓度为0.2 mmol/L,25℃分别诱导4、8、12、16、20和24 h。如图7所示,加入诱导16 h左右的比酶活达到最高值,之后酶活变化幅度很小。这表明到达一定的表达量以后,再过量的外源酶蛋白会对菌体产生负荷,部分会被菌体所水解,过度延长诱导时间不具实用意义[16]。
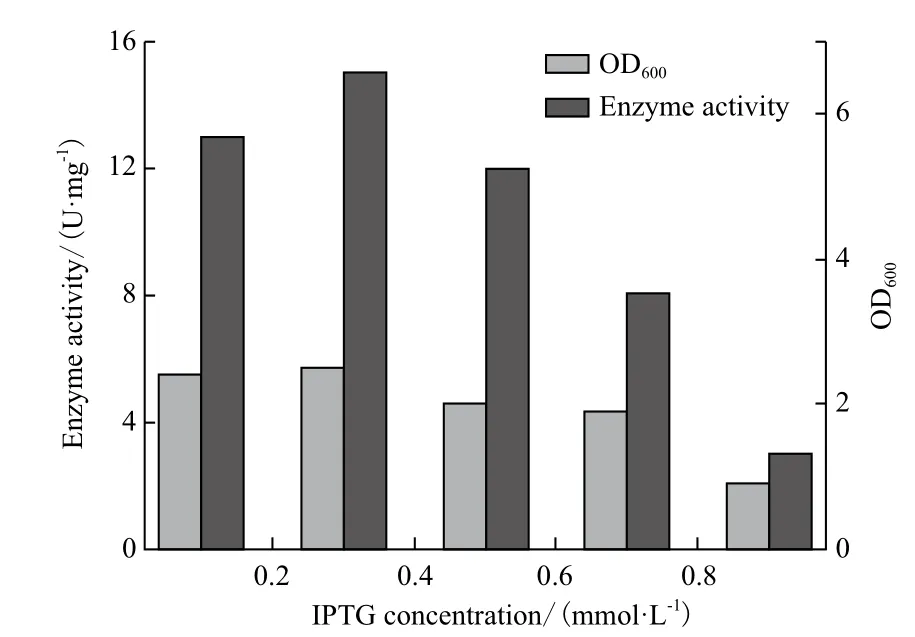
图6 IPTG浓度的优化

图7 诱导时间优化
经过一系列的优化后,在200 mL装液量的摇瓶中放大培养,使用25℃培养温度,终浓度为0.2 mmol/的IPTG诱导表达16 h,重组菌的活力可达到15 U/mg,比优化前提高了5倍。
3 讨论
腈水解酶在医药、化工和有机合成等方面具有十分广泛的应用,目前国内有关该方面的研究还处在较落后水平,且有关研究还多集中在扁桃酸、烟酸等一些比较基础的研究层面[17],有关化学选择性和立体选择性相结合的腈水解酶筛选更是鲜有耳闻,有关的酶蛋白克隆表达和分子改造等相关研究较少。在国外特别是欧洲和美国等国家,有关研究可追溯到20世纪80到90年代[18]。
pET表达系统具有乳糖诱导性能,通过α互补作用,含有该质粒菌株能利用乳糖的类似物IPTG来高表达目的蛋白[19,20]。虽然有敏捷食酸菌的腈水解酶基因克隆的相关报道[21],但有关诱导表达和产酶的优化研究还未见报道。本研究切除了基因自带的启动子,采用pET-28自带的T7强启动子对其进行表达,选择了能利于稀有密码子表达的大肠杆菌Rosetta株作为宿主菌。在优化了一系列表达条件下,酶的表达量和活力均有提高,而质粒所带的His-tag标签对于蛋白纯化十分有利。在酶活测定方面,本试验测量量化到了毫克比酶活,并使用苯酚硫酸盐显色法,测定更加快捷和精确,有助于研究转化反应酶用量的控制。具有化学选择性的脂肪族腈水解酶工程菌的成功构建,为使用该菌株催化相关底物的研究,以及相关分子改造奠定了实验基础。
4 结论
本研究以具有化学选择的敏捷食酸菌腈水解酶DNA作为模板,PCR扩增获得目的片段,测序验证所克隆的目的片段,选用pET-28a质粒作为表达载体构建表达重组质粒并转化。对重组菌株进行诱导表达,优化诱导条件,结果显示经过优化后比酶活大幅度提高,优化后的酶比活力高达15 U/mg,比未优化的提高了5倍。
[1]徐建妙, 郑裕国, 沈寅初. 腈水解酶的来源, 结构, 作用机制及其应用[J]. 微生物学通报, 2005, 32(5):141-146.
[2] Pace HC, Brenner C. The nitrilase superfamily:classification, structure and function[J]. Genome Biol, 2001, 2(1):1-9.
[3]Gong JS, Lu ZM, Li H, et al. Nitrilases in nitrile biocatalysis: recent progress and forthcoming research[J]. Microb Cell Fact, 2012,11: 142.
[4]Brenner C. Catalysis in the nitrilase superfamily[J]. Current Opinion in Structural Biology, 2002, 12(6):775-782.
[5]何玉财, 许建和. 腈水解酶在羧酸合成中的研究进展[J]. 生物加工过程, 2009, 7(1):7-12.
[6]Panova A, Mersinger LJ, Liu Q, et al. Chemoenzymatic synthesis of glycolic acid[J]. Advanced Synthesis & Catalysis, 2007, 349(8-9):1462-1474.
[7]许建和, 何玉财, 张志钧. 一种产碱杆菌的培养及其于腈水解制备羟基乙酸的方法:中国, CN1011386933A[P]. 2008.
[8] Baum S, van Rantwijk F, Stolz A. Application of a Recombinant Escherichia coli whole-cell catalyst expressing hydroxynitrile lyase and nitrilase activities in ionic liquids for the production of(S)-mandelic acid and(S)-mandeloamide[J]. Advanced Synthesis and Catalysis, 2012, 354(1):113-122.
[9] 王学东, 李桂南, 李明阳. 浑球红细菌 Rhodobacter sphaeroides LHS-305 腈水解酶基因的克隆及表达[J]. 重庆理工大学学报:自然科学版, 2012, 26(6):24-28.
[10] Xie Z, Feng J, Garcia E, et al. Cloning and optimization of a nitrilase for the synthesis of(3-S-3-cyano-5-methyl hexanoic acid[J]. Journal of Molecular Catalysis B:Enzymatic, 2006, 41(3):75-80.
[11]吴中柳, 李祖义. 氰基水解酶在有机合成中的应用[J]. 有机化学, 2001, 21(1):25-32.
[12] Gavagan JE, DiCosimo R, Eisenberg A, et al. A Gram-negative bacterium producing a heat-stable nitrilase highly active on aliphatic dinitriles[J]. Applied Microbiology and Biotechnology,1999, 52(5):654-659.
[13] 李宝齐, 邓英杰, 杨静文. 苯酚-次氯酸盐法测定空白脂质体透析液中铵离子浓度[J]. 中国药剂学杂志, 2006(4):181-185.
[14]Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding[J]. Analytical Biochemistry, 1976, 72(1):248-254.
[15] 罗惠霞, 李敏, 王玉炯. 包涵体蛋白复性的几种方法[J]. 生物技术通报, 2007(5):96-98.
[16] 李永仙, 郑飞云, 李崎, 等. 重组大肠杆菌 BL21(DE3)-pET28a(+)-bgl 诱导表达 β-葡聚糖酶的条件优化[J]. 食品与生物技术学报, 2009, 28(2):250-255.
[17]何玉财, 周琼, 张跃, 等. 一株烟腈水解酶菌株的筛选及其催化特性初步研究[J]. 化工进展, 2012, 30(12):2714-2718.
[18]Schmidt RC, Müller A, Hain R, et al. Transgenic tobacco plants expressing the Arabidopsis thaliana nitrilase II enzyme[J]. The Plant Journal, 1996, 9(5):683-691.
[19] 陈卫, 葛佳佳, 张灏, 等. 半乳糖苷酶基因在大肠杆菌中过量表达及 IPTG 诱导条件[J]. 无锡轻工大学学报, 2002, 21(5):492-495.
[20] 吴一凡, 张双全. 乳糖诱导 pET 载体表达重组蛋白的研究[J].南京师大学报:自然科学版, 2002, 25(1):89-93.
[21] Chauhan S, Wu S, Blumerman S, et al. Purification, cloning, sequencing and over-expression in Escherichia coli of a regioselective aliphatic nitrilase from Acidovorax facilis 72W[J]. Applied Microbiology and Biotechnology, 2003, 61(2):118-122.
(责任编辑 李楠)
Expression of Nitrilase Gene from Acidovorax facilis in Escherichia coli
Guan Qiwang Ma Jiangfeng He Aiyong Jiang Min Gong Changbin
(State Key Laboratory of Materials-Oriented Chemical Engineering,College of Biotechnology and Pharmaceutical Engineering,Nanjing Tech University,Nanjing 211816)
Plasmid containing Acidovorax facilis gene, was used as a template, and length of 1 080 bp nitrilase(Nitrilase)gene fragment was obtained by PCR. Expressed genes were sequenced and compared to gene sequences found in GenBank. There are 2 bp mutation was found, which can not affect the expression and characterization of the enzyme. The recombinant plasmid was transformed into the Escherichia coli Rosetta(DE3)competent cell, which was induced IPTG for strain- expression. SDS-PAGE analysis was used and showed that the protein molecular weight was about 41.28 kD. Enzyme activity analysis showed that the specific activity of the supernatant was 3 U/mg. Induction conditions were optimized, including induction temperature, IPTG concentration, induction time and a series of conditions. Under optimal conditions and with the expansion of the culture volume, the specific activity can up to 15 U/mg, and activity increased by about five times.
nitrilase;Acidovorax facilis;Escherichia coli;clone
10.13560/j.cnki.biotech.bull.1985.2015.01.027
2014-06-12
国家自然科学基金项目(21076105),国家重点基础研究发展计划(“973”计划)(2009CB724701),江苏高校优势学科建设工程项目
管启旺,男,硕士,研究方向:工业微生物;E-mail:819468208@qq.com
马江锋;E-mail:bioengine@njut.edu.cn
