临床试验伦理委员会初始审查中的主要问题
2015-09-21曾代文杨友松韩盛玺
全 婷,曾代文,杨友松,邹 静,韩盛玺
(四川省医学科学院/四川省人民医院药物临床试验机构办公室,四川 成都 610072,quanting83@gmai1.com)
临床试验伦理委员会初始审查中的主要问题
全婷,曾代文*,杨友松,邹静,韩盛玺
(四川省医学科学院/四川省人民医院药物临床试验机构办公室,四川 成都 610072,quanting83@gmai1.com)
目的 了解伦理审查的质量关系到受试者的权益是否得到有效保护。方法 通过《药物临床试验伦理审查工作指导原则》出台后两年该伦理委员会的审查工作的分析,总结了目前伦理委员会审查中的方案和知情同意书的常见问题。结果 该伦理委员会通过召开伦理例会的形式审查了94个新立项的药物/器械临床试验项目,其中29个项目的初始审查结论为同意,会议审查一次性通过率仅有31%。其中,方案中出现最频繁的问题包括:试验背景和立题依据、试验设计、试验风险与受益三方面;知情同意书的主要问题集中于知情同意书的内容、语言和签署页的设计。结论 受试者的保护仍需研究者、申办者、药物临床试验机构及伦理委员会能力的共同提高。
伦理委员会;临床试验;伦理审查;初始审查
1 药物/器械临床试验初始伦理审查中的常见问题
近两年间,笔者所在单位伦理委员会通过召开伦理例会的形式审查了94个新立项的药物/器械临床试验项目,其中29个项目的初始审查结论为同意,其余均有针对方案或知情同意书的具体修改意见,会议审查一次性通过率仅有31%。其中,方案中出现最频繁的问题包括:试验背景和立题依据、试验设计、试验风险与受益三方面;知情同意书的主要问题集中于知情同意书的内容、语言和签署方面(详见表1)。
2 方案中的主要伦理问题分析
2.1研究背景和立题依据
以人为研究对象决定了临床试验的特殊性,《赫尔辛基宣言》指出:涉及人体受试者的研究必须符合公认的科学原则,并以对科学文献、其他相关资料、充分的实验室研究、适当的动物实验的充分了解为基础。[1]ICH-GCP要求药物临床试验必须有充分的非临床的和临床的信息支持。[2]
缺乏充分的基础研究,直接将试验产品用于人体试验,或是重复已有明确结论的研究,浪费人力物力财力的同时,不但让受试者毫无收益,而且会将受试者暴露在不必要的风险之中。例如,对于已有的同类产品,研究背景中应明确新申报的医疗器械相对于已上市销售的产品是否具有优势,性能、价格上相对于进口产品是否具有优势。对于药物或器械上市后临床评价研究,伦理委员会应关注研究的科学和社会价值,特别是某些针对于高值耗材的研究,是否单纯以市场推广为目的。
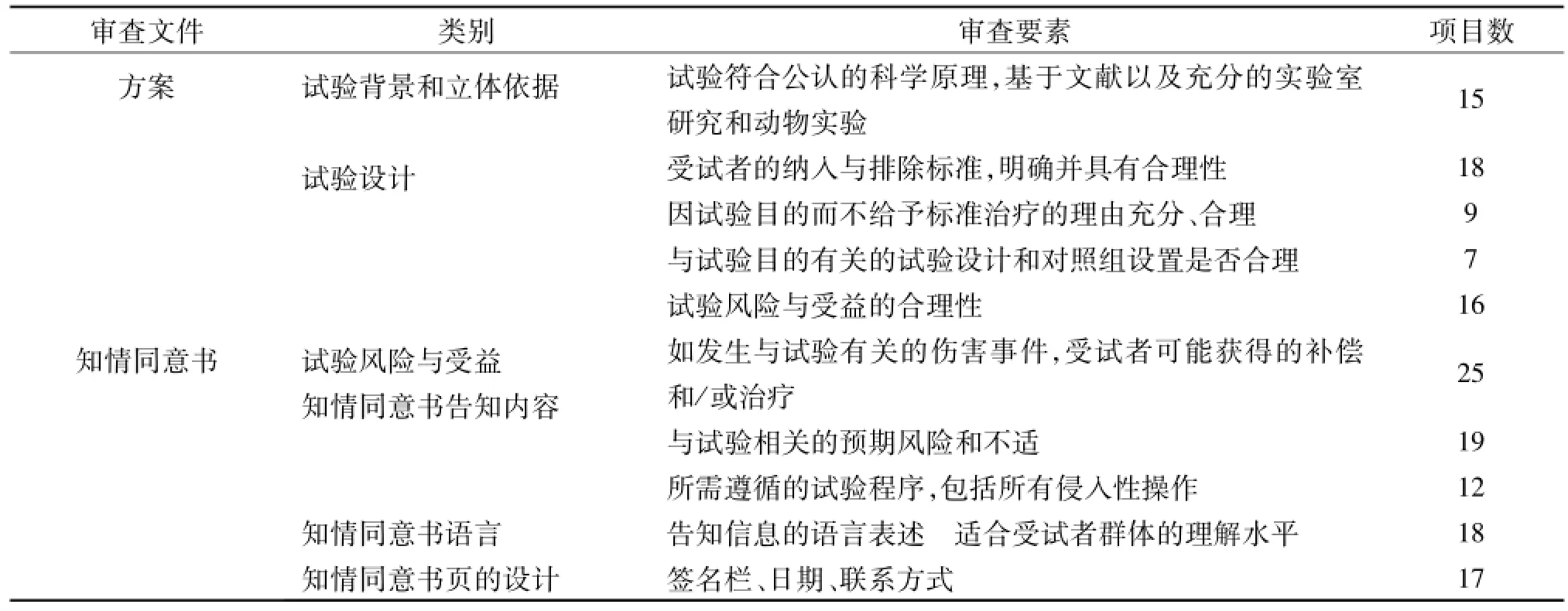
表1 方案和知情同意书中的主要问题
2.2试验设计的合理性
试验设计如果达不到临床研究的最终目的,受试者对于科学研究的奉献也是徒劳的。例如,为评价降糖药物的有效性,未进行糖化血红蛋白的检测,只检测空腹和餐后2h血糖。但空腹血糖和餐后2h血糖属短期血糖检测方法,反映的是即时血糖水平且影响因素较多、波动较大,而糖化血红蛋白和糖化血清蛋白均属于非酶促反应产物的糖化蛋白,其稳定性好,对于判断疾病预后和药物疗效更具说服力。[3]为评价某药物的安全性,前期研究发现药物可能对血清磷产生影响,临床试验方案却未对受试者进行血清磷和骨密度检测。这样的设计最终达不到评价药物疗效性及安全性的目的。
2.2.1入选排除标准。
入选排除标准主要是受试者的选择问题。临床试验应该选择目标适应症人群进行研究,同时根据对受试产品的有效性和安全性的认知程度,对存在高风险因素的患者,以及可能影响最终疗效和安全性评价的各类复杂而难以控制的因素加以限制。入选排除标准还应考虑到不给受试者增加额外的风险。例如,在一项研究磺酰脲类联合二甲双胍治疗血糖控制不佳的2型糖尿病患者加用某试验药物的安全性和有效性研究中,未将胰腺炎患者排除在外。而2010年美国食品药品监督管理局(FDA)针对这个问题已修改了该试验药物的说明书,建议医务工作者在开始用该药或增加该药剂量时,应严密监测胰腺炎的风险,一旦怀疑发生胰腺炎应立即停药。[4]此研究中若纳入了合并胰腺炎的受试者,将大大增加受试者的研究风险。
2.2.2对照组的选择。
在试验对照的选择方面常常出现以下问题:临床试验不设置对照组、所选的对照药物缺乏证明其疗效的数据、安慰剂对照使用不合理等。使用安慰剂、空白对照及效果较差的干预措施时,必须证明其必要性。
不科学的研究方案设计肯定是违背伦理的。例如,一项研究要求在受试者服药1、2、4、8、12、16、20、24、28、32及36周抽血检测药物的血药谷浓度,但要求研究者提供检测的为血清,血药浓度检测应该使用的是全血,如果研究者向实验室提供血清,不但无法达到检测血药浓度的目的,反而会让受试者反复抽血,暴露于不必要的风险之中。科学的设计可能以最简单有效的方式直接获得研究结果,但以科学的名义简单地进行人体研究,可能会造成极为严重的后果。
2.3受试者的风险和受益
受试者的风险和受益是伦理委员会关注的重点。临床研究具有不确定性,有时受试者可能因为参加研究获得了研究者更多的关怀,或免费获得了有确切疗效的药物,但仍有很多受试者可能不会直接受益。伦理委员会需要考虑研究是否具有社会价值,在能达到试验目的的前提下受试者风险是否已降到最低,受试者参加研究的风险受益比是否合理。例如,某化疗药物预计能够延长肿瘤患者的无病生存时间为3个月,但其预期的不良反应可能使受试者后期的生存质量大大降低,此类研究的风险获益比值得考量。又如,针对健康志愿者开展的I期临床试验,受试者本人在研究中不能获益,但I期临床研究是新药研发中的一个不可或缺的环节。为降低受试者的风险,除了科学设计研究方案之外,研究者还会给予受试者一定的经济补偿。补偿太少于受试者不合理,补偿过多又有诱导嫌疑,从而催生出了“专业试药人”。
3 知情同意书中主要伦理问题分析
3.1告知内容不充分
知情同意书故意回避发生试验相关损害的补偿或赔偿问题,或单纯地描述为:因为试验药物导致的相关损害,由申办方承担相应的治疗费用和补偿。例如:“对于标准化疗方案出现相同类别、频率及程度相似或相同的不良事件或可预见的不良事件……申办方不予进行补偿”。根据《药物临床试验质量管理规范》,受试者签署知情同意书后即视为开始加入试验,此后受试者按试验的全部流程配合操作和观察,与此相关的所有损害责任申办者都应当承担。受试者有权利知道具体的赔偿和补偿信息。不能将研究的风险与标准治疗方案进行对比,以此划定申办者的责任范围。
有的知情同意书关于试验相关的预期风险和不适描述不准确,或直接阐述发生不良反应的概率极低。使受试者不能充分了解试验风险,难以做出正确的判断。这种操作一方面违背了医学研究的基本伦理道德,另一方面也不利于试验开展。一旦发生不良反应,极可能对受试者造成损害或发生医患纠纷。
有的知情同意书告知内容信息不充分,研究的基本流程没有清楚的阐述。这违背了充分告知的伦理原则,也不利于受试者对试验的充分配合。例如,知情同意书与研究方案对操作流程不一致,研究方案要求每1年做一次B超监测,知情同意书告知每半年做一次监测。有的药物上市后评价试验产品免费,对照产品需要自费承担,还需要受试者进行额外的实验室检测,但在知情同意书中均没有明确地告知,最终造成受试者利益受到损害,权益得不到保障。
3.2知情同意书的语言
向受试者提供的试验信息资料必须完整易懂,有的知情同意书关于试验的描述太过专业,易造成普通受试者不能充分理解试验信息。如果有针对未成年人的知情同意书,还应以符合受试者年龄和理解能力的语言来介绍和解释研究内容。知情同意书应避免不恰当或明显诱导性语言,例如某止咳贴临床试验的知情同意书提到“西药有副作用,中药只会给您带来不便”。
3.3知情同意书签署的设计
知情同意书由法定代理人或监护人代受试者签字时,需要注明与受试者的关系。知情同意书的告知页最好与签署页分开,当试验受试对象为未成年人时,除需要监护人签字外,还应征得其本人同意。
4 讨论
《药物临床试验质量管理规范》要求研究者与申办者一起完善临床试验方案,[5]申办者是临床试验的发起者和最大直接获益者,许多申办者会聘请合同研究组织(CRO)协助撰写方案,研究者参与讨论,但由于CRO公司的专业团队水平参差不齐,许多方案从总体设计、结构到具体的文字都可能存在问题。国内的医疗器械临床试验一般投入较少,一般由申办者直接与研究者讨论撰写方案,许多医疗器械的厂家从未参与过临床试验或者好几年前做过一些产品的临床试验,对于专业的临床研究方案撰写缺乏经验。由于医疗器械的多样性,临床试验涉及的专业范围较广,有的专业可能从未进行过注册临床研究,研究者也缺乏撰写临床试验方案的经验。综上所述,造成研究方案和知情同意书的撰写在科学性和伦理性方面都可能存在问题。这对于伦理委员会的审查能力也是个极大的考验。
多年以来,我国伦理委员会多重视项目方案和知情同意书的审查,对于临床试验项目的跟踪和结题审查经验尚不足。尽管不少伦理委员会已要求研究者需向伦理委员会提交严重不良事件报告、违背方案报告、研究进展报告和结题报告等,但不少研究者和申办者的重视程度还不够,或还未养成持续向伦理委员会报告的习惯,需要药物临床试验机构和伦理委员会持续开展伦理学培训及督促研究者履行职责。
伦理委员会审查能力的提高是一个长期积累的过程,不但需要有较强的专业技术知识,还需要有丰富的审查经验及对伦理学理论的不断实践。伦理委员会的成员也应该不断更新现有的知识,才能实现对方案和知情同意书科学性和伦理性的有效审查,最终保护受试者的安全和权益。
[1] 世界医学会.赫尔辛基宣言[Z].2013.
[2] ICH.E6 Guide1ine for Good C1inica1Practice[EB/ OL].http://www.ich.org/fi1eadmin/Pub1ic-Web -Site/ICH-Products/Guide1ines/Efficacy/E6-R1/ Step4/E6-R1--Guide1ine.pdf.
[3] 唐健元,马莉.糖化血红蛋白和糖化血清蛋白在糖尿病中药新药临床研究设计中的作用[J].中药新药与临床药理,2007,18(2):162-164.
[4] 蒋彦章.西格列汀:修订该药处方及其致急性胰腺炎信息[J].药物流行病学杂志,2011,20(3):165.
[5] 国家食品药品监督管理局.药物临床试验质量管理规范[Z].2003.
〔修回日期 2014-12-26〕
〔编 辑 吉鹏程〕
Common Issues in the Initial Review of the Clinical Trial Ethics Comm ittee
QUAN Ting,ZENG Daiwen,YANG Yousong,ZOU Jing,HAN Shengxi
(Sichuan Academy Of Medical Sciences/Office of Clinical Trial Institution,Sichuan Provincial People's Hospital,Chengdu 610072,China,E-mail:quanting83@gmail.com)
Ob jective:To ana1yze that if the qua1ity of ethics review is c1ose1y re1ated to the protection of human subjects'right and interest.Methods:This artic1e has ana1yzed a11 the issues raised by 1oca1Ethics Committee in the process of review in recent two years since″guide1ine of ethica1 review of drug c1inica1 tria1s″was pub1ished,summed up themost common prob1emsoccurred in protoco1sand informed consents.Results:Tota194 new drug or medica1device c1inica1tria1projectswere reviewed by the 1oca1ethics committee,amongwhich 29 projectswere approved through regu1ar fu11boardmeeting,the approva1rate in the initia1review was31%.Themost common prob-1ems in protoco1s inc1ude:the research backgrounds,design,and risk-benefit ratio;Main issues raised on informed consent focused on the contents,1anguage and signature terms.Conclusions:The protection of subjects needsmore improvement of capabi1ity of investigator,sponsor,drug c1inica1 tria1institution and the ethics committee.
Ethics Committee;C1inica1Tria1;Ethica1Review;Initia1Review
R-052
A
1001-8565(2015)01-0047-03
,E-mai1:zdw.11@163.com
2014-08-06〕
