连翘配方颗粒高效液相色谱指纹图谱
2015-09-15梁俊黄良永杨光义
梁俊,黄良永,杨光义
(湖北医药学院附属太和医院药学部,十堰 442000)
连翘为木犀科植物连翘[Forsythia suspensa(Thunb.)Vahl]的干燥果实,具有清热解毒、消肿散结、疏散风热的功效。用于痈疽,瘰疬,乳痈,丹毒,风热感冒,温病初起,温热入营,高热烦渴,神昏发斑,热淋涩痛[1]。主要含有挥发性成分、苯乙醇苷类、木脂素类和三萜类物质[2],以连翘酯苷A和连翘苷的含量较高。现代研究表明,连翘酯苷A 对认知障碍及短暂性脑缺血的神经具有保护作用,故具有改善学习记忆障碍、舒张血管、抗氧化、抗菌、抗感染、解热、抗DNA损伤、防护耳毒性等药理作用[3]。连翘配方颗粒是以连翘为原料,经水提取、浓缩、喷雾干燥而制成的颗粒,有文献报道采用高效液相色谱(HPLC)法测定连翘配方颗粒中连翘苷含量[4-5],但还没有统一的质量标准。笔者应用HPLC-梯度洗脱法,建立了连翘配方颗粒的HPLC指纹图谱,以便为建立质量标准提供参考。
1 仪器与试药
1.1 仪器 DGLC双三元高效液相色谱仪(美国Thermo公司,双三元梯度泵、WPS3000自动进样器、AccelaPDA二极管阵列检测器、TCC柱温箱);Chromeleon色谱工作站;Waters SunFire C18色谱柱(美国 WATERS 公司,4.6 mm ×150 mm,5 μm);520H 超声清洗机(南京熊猫电子集团,250 W,40 kHz);AUW120D电子分析天平(日本岛津公司,感量:十万分之一)。
1.2 试药 甲醇为色谱纯(美国赛默飞公司),水为超纯水,其他试剂均为分析纯。连翘酯苷A对照品(批号:111810-201304,含量:94.3%)、连翘苷对照品(批号:110821-200711,含量:98.9%)均由中国食品药品检定研究院提供。连翘配方颗粒(广东深圳某医药股份公司生产,每袋1 g,相当于连翘药材10 g,批号:10041002S-S1、1011001S-S2、1103001S-S3、1105002SS4、 1107003S-S5、 1110001S-S6、 1203001S-S7、1302002S-S8、1307003S-S9、1311001S-S10)。
2 方法与结果
2.1 测定方法
2.1.1 色谱条件 Waters SunFire C18色谱柱(4.6 mm ×250 mm,5 μm);流动相 A 为乙腈,流动相B为0.4%醋酸溶液,按表1程序进行线性梯度洗脱;流速:1.0 mL·min-1。柱温:30 ℃;检测波长:277 nm;进样 10 μL[1]。

表1 流动相线性梯度表Tab.1 Linear gradient table of mobile phase %
2.1.2 样品溶液的制备 混合对照品溶液的制备:精密称取连翘酯苷A对照品21.70 mg,置25 mL棕色量瓶,加甲醇溶液超声溶解并稀释至刻度,摇匀,得储备液(每毫升含连翘酯苷A 868.0 μg);另精密称取连翘苷对照品20.60 mg置50 mL棕色量瓶,加甲醇溶液超声溶解并稀释至刻度,摇匀;即贮备液(含连翘苷412.0 μg)。再分别精密量取连翘酯苷A和连翘苷贮备液2.5 mL和0.5 mL置同一10 mL量瓶中,用甲醇溶液稀释至刻度,即得混合对照品溶液(每毫升含连翘酯苷 A217.0 μg,连翘苷 20.60 μg)。
供试品溶液的制备:取连翘配方颗粒研碎,过五号筛,精密称取0.5 g,置具塞锥形瓶,精密加入70%甲醇溶液15 mL,密塞,称定质量,超声处理(功率250 W,频率40 kHz)30 min,放冷,再称定质量,用70%甲醇溶液补足减失的质量,摇匀,滤过。取续滤液,即得。
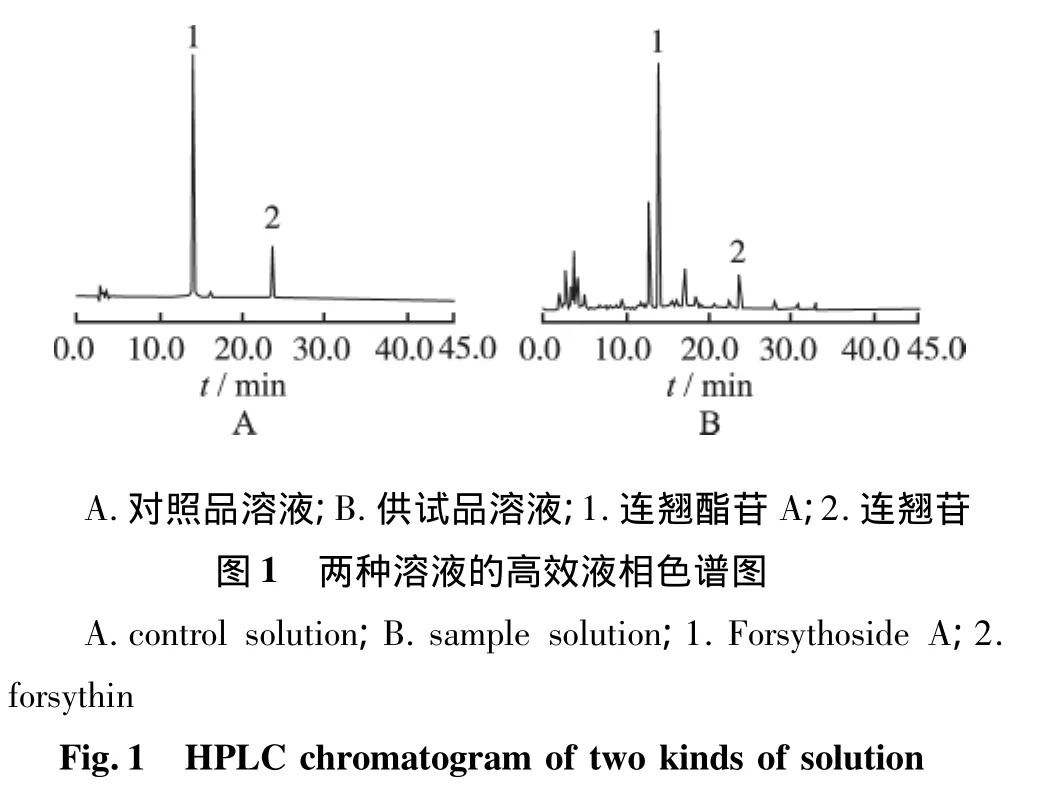
2.1.3 系统适用性实验 精密取混合对照品溶液、供试品溶液各10 μL,注入液相色谱仪,在“2.1.1”项色谱条件下,连翘配方颗粒的供试品溶液在对照品色谱相同的位置有连翘酯苷A和连翘苷的峰,二成分峰与其相邻峰完全分离,理论板数以连翘酯苷A峰计均>18 000。二成分峰纯度经PDA检测器扫描对比符合度99.7%,说明和对照品的峰完全一致。结果见色谱图1。
2.2 指纹图谱方法学考察
2.2.1 精密度实验 取同一供试品溶液(批号:1311001S-S10),按“2.1.1”项色谱条件连续进样 5 次,每次10 μL,记录色谱图,测定主要共有峰的相对保留时间和相对峰面积,结果主要共有峰13个,1~13号峰相对保留时间的RSD均<0.10%,相对峰面积的RSD均<0.44%,符合指纹图谱要求,表明仪器精密度好。
2.2.2 重复性实验 取同一样品(批号:1311001SS10)6份,按“2.1.2”项方法制备供试品溶液,按“2.1.1”项方法进样测定并分别测定其色谱图,结果其相对保留时间稳定,主要共有峰1~13号峰相对保留时间RSD<0.15%,相对峰面积RSD均<0.90%,说明本方法重复性较好。
2.2.3 稳定性实验 取同一样品(批号:1311001SS10)溶液在0,4,12,24 h 分别按“2.1.1”项方法进样测定并分别记录其色谱图,测定主要共有峰的相对保留时间和相对峰面积,结果主要共有峰13个,1~13号峰相对保留时间RSD均<0.10%,相对峰面积RSD均<0.48%,结果表明供试品溶液室温下0~24 h内成分保持稳定,能满足指纹图谱要求。
2.3 已知成分的鉴别和参比峰的确认 供试品溶液和对照品溶液在“2.1.1”项色谱条件下得到的色谱图,通过色谱峰的保留时间和二极管阵列检测器的符合度检查比对,对指纹图谱中相应色谱峰进行确认,结果显示6号峰为连翘酯苷A;10号峰为连翘苷。其中6号连翘酯苷A峰的保留时间居中,峰面积值较大且稳定,所以选择6号峰为参比峰(图2)。
2.4 指纹图谱的建立和相似度评价 取10批连翘配方颗粒样品,分别按“2.1.2”项供试品的制备方法处理,在“2.1.1”项色谱条件下,测定其色谱图。
利用中药指纹图谱相似度评价系统(2004 A版)软件,采用自动匹配法生成共有模式对照图谱R并分析各样品之间共有峰和相似度,确定共有峰。结果发现10批供试品共有13个共有峰,见图2和图3。各特征图谱与共有模式对照图谱R相似度见表2。以选定的6号峰连翘酯苷A峰为参比峰,设定其相对保留时间和峰面积为1,计算各共有峰相对它的相对保留时间和相对峰面积,结果见表3和表4。
3 讨论
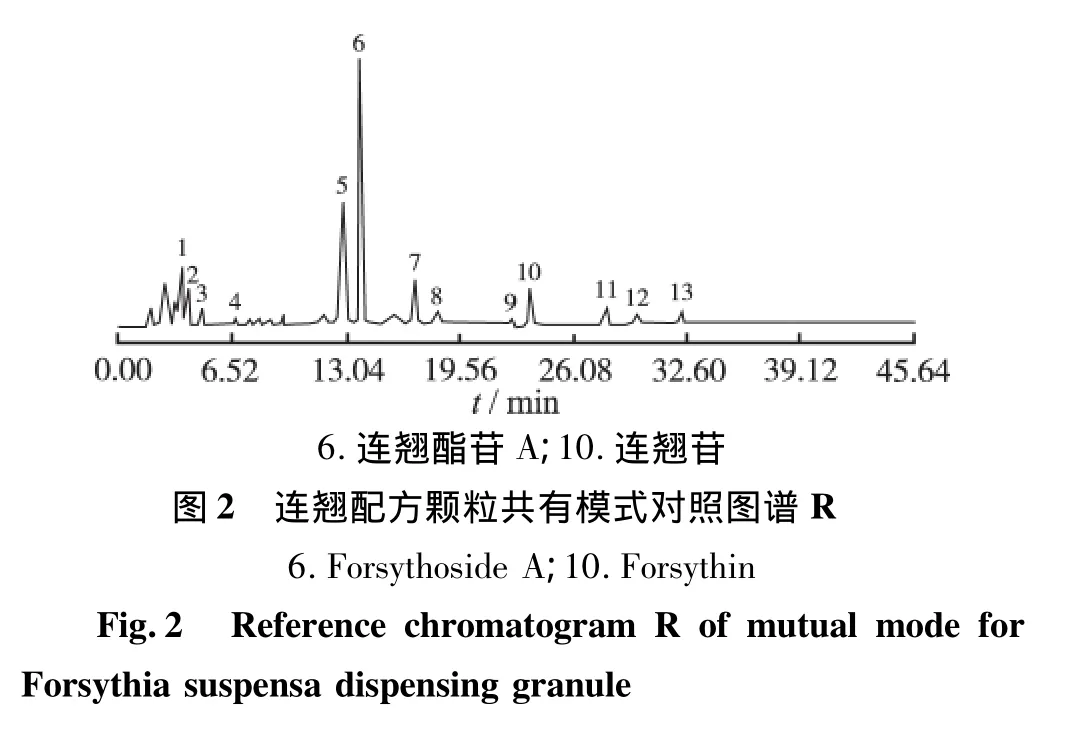
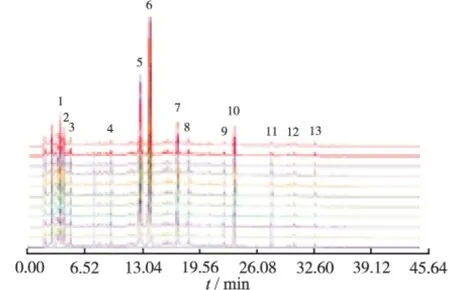
图3 10批样品的HPLC指纹图谱相似度比较Fig.3 Similarity comparison of HPLC fingerprints among ten batches of samples
在筛选色谱条件时,流动相采用《中华人民共和国药典》2010年版连翘项下的含量测定方法,需要使用两个色谱条件分别测定连翘酯苷A和连翘苷的含量,对于HPLC指纹图谱的测定不太适应;笔者在本实验中采用乙腈-0.4%醋酸溶液进行梯度洗脱,流动相极性由大到小,使连翘酯苷A和连翘苷与其相邻色谱峰完全分离,可准确测定二成分的含量,取得满意结果。
建立样品的制备方法时,用不同比例的甲醇溶液(20%,40%,50%,70%,100%)直接超声提取,经过比较,采用《中华人民共和国药典》2010年版的方法,用70%乙醇溶液超声提取,两成分的回收率和重复性最好。
实验中发现,10批样品的HPLC指纹图谱中,13个共有峰的相似度均>0.999,说明连翘配方颗粒的HPLC指纹图谱稳定可靠;各批号样品间13个共有指纹峰的相对保留时间比较吻合(RSD<0.31%);相对峰面积波动较小(RSD<10%),说明该公司生产的不同批次颗粒的成分含量差异较小,生产颗粒的原药材来源一致,质量稳定。实验结果表明,利用HPLC色谱指纹图谱来控制连翘配方颗粒的质量切实可行,该法为该产品的质量控制提供了可行方法。
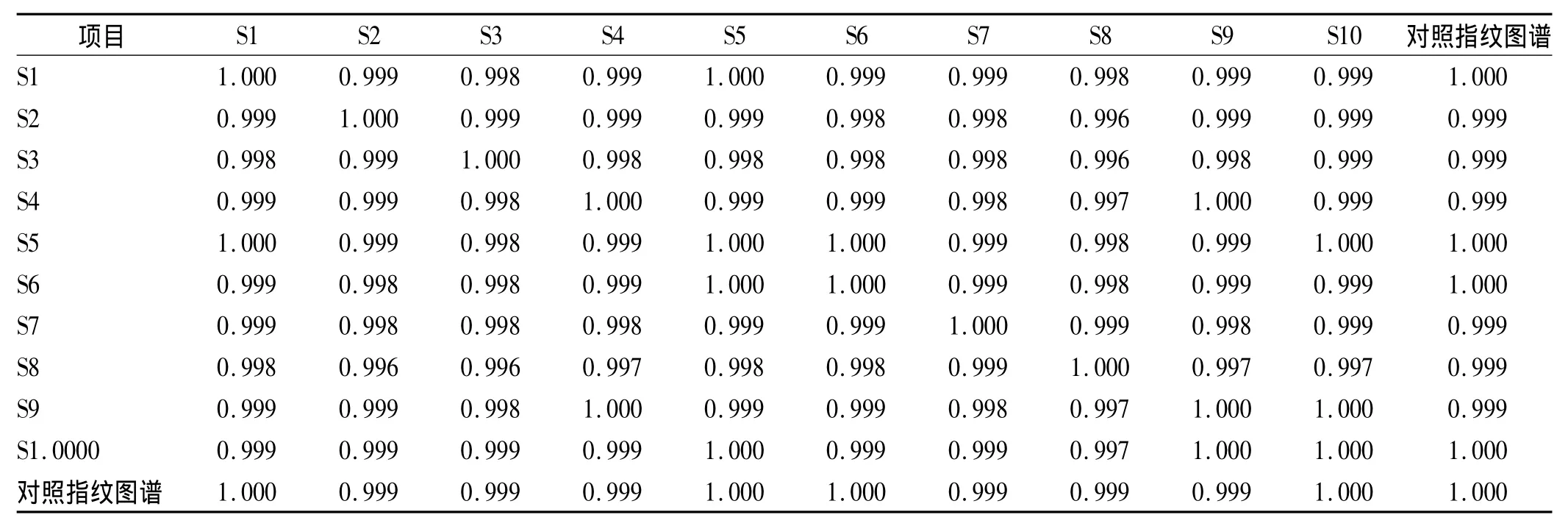
表2 样品特征图谱与共有模式对照图谱R的相似度Tab.2 Similarity between characteristic chromatogram and reference chromatogram R of mutual mode

表3 HPLC指纹图谱共有峰相对保留时间Tab.3 Relative retention time of common peaks of HPLC fingerprints

表4 特征图谱共有峰相对峰面积Tab.4 Relative peak area of common peaks of characteristic chromatogram
[1]国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2010:159.
[2]孟祥乐,李俊平,李丹,等.连翘的化学成分及其药理活性研究进展[J].中国药房,2010,21(43):4117 -4118.
[3]吴国友.连翘药理作用研究进展[J].中医学报,2013,28(185):1508-1509.
[4]黄蓓蓓,贺帅.微乳液相色谱法同时测定注射用双黄连冻干粉中三组分的含量[J].医药导报,2013,32(1):92 -95.
[5]赵维娟,刘静,边佳明.复方连翘皮康乳膏中3种有效成分的含量测定[J].医药导报,2012,31(7):926 -928.
