吲达帕胺缓释片有关物质测定
2015-09-15袁春平张文芳梁华娟吴志权
袁春平,张文芳,梁华娟,吴志权
(广东环球制药有限公司,佛山 528305)
吲达帕胺属噻嗪类利尿药,是一种有效的作用强的利尿降压药物[1]。该缓释片治疗剂量低,疗效明显,体内积蓄少,适合高血压患者长期应用[2-3]。为更好地控制产品质量,将杂质可能带来的安全性隐患降至最小,本研究建立两种高效液相色谱(HPLC)法测定吲达帕胺缓释片的有关物质,方法一采用外标法测定吲达帕胺缓释片中4-氯-3-氨磺酰胺苯甲酸(起始原料),加校正因子的主成分自身对照法测定杂质B(脱氢吲达帕胺,主要的降解杂质),并对其他单杂质及总杂质进行控制;虽然方法一能良好分离杂质A(N-亚硝基-2-甲基吲哚啉),但由于进样量和检测限的限制,需建立方法二测定杂质A。报道如下。
1 仪器与试药
1.1 仪器 岛津LC-10AT高效液相色谱仪;岛津LC-20AT高效液相色谱仪,SPD-M20A二极管阵列检测器。AL104型、MS105DU型电子天平[梅特勒-托利多仪器(上海)有限公司,分别为万分之一和十万分之一天平]。
1.2 试药 吲达帕胺缓释片(广东环球制药有限公司研制,规格:每片 1.5 mg,批号:110301,110302,110303),吲达帕胺对照品(中国食品药品检定研究院,批号:100257-200903);吲达帕胺杂质 B对照品(USP,批号:1338812),4-氯-3-氨磺酰胺苯甲酸(东京化成工业株式会社,批号:SQ5AD-EJ),吲达帕胺杂质A对照品(EP,批号:2.1);乙腈、四氢呋喃、冰醋酸、三乙胺为色谱纯,超纯水为自制,其他试剂均为分析纯。
2 方法与结果
2.1 4-氯-3-氨磺酰胺苯甲酸和杂质B的测定(方法一)
2.1.1 色谱条件 Welch Ultimate C18柱(250 mm ×4.6 mm,5 μm);流动相:水∶乙腈∶2-丁醇∶三乙胺∶十二烷基硫酸钠溶液(取十二烷基硫酸钠5 g,加冰醋酸3 mL,加水100 mL,摇匀)=690∶310∶20∶10∶6(用磷酸调节pH至3.0);检测波长240 nm,流速1.6 mL·min-1,柱温 30 ℃,进样量 20 μL。
2.1.2 溶液的配制
2.1.2.1 供试品溶液 取本品适量研细,取细粉适量(相当于吲达帕胺15 mg),精密称定,置50 mL量瓶中,加乙醇适量充分振摇使溶解并稀释至刻度摇匀。离心,精密量取上清液2 mL,置5 mL量瓶中,加流动相稀释至刻度,摇匀,即得。
2.1.2.2 供试品-自身对照溶液 精密量取供试品溶液1 mL,置100 mL量瓶中,加流动相稀释至刻度,摇匀,即得。
2.1.2.3 4-氯-3-氨磺酰胺苯甲酸对照品溶液 取4-氯-3-氨磺酰胺苯甲酸对照品约12.5 mg,精密称定,置100 mL量瓶中,加乙醇溶解并稀释至刻度,摇匀。精密量取2 mL,置100 mL量瓶中,加乙醇稀释至刻度,摇匀,作为贮备溶液,精密量取贮备溶液2.4 mL,置10 mL量瓶中,加流动相稀释至刻度,摇匀,即得。
2.1.2.4 系统适用性溶液 取杂质B、4-氯-3-氨磺酰胺苯甲酸对照品各约1.2 mg,精密称定,置同一个100 mL量瓶中,加乙醇溶解并稀释至刻度,摇匀,精密量取2 mL,置10 mL量瓶中,加乙醇稀释至刻度,摇匀,作为杂质贮备溶液;另取杂质A对照品约1.2 mg,精密称定,置100 mL量瓶中,加乙醇溶解并稀释至刻度,摇匀,作为杂质A贮备溶液;精密称取吲达帕胺对照品约3 mg,置10 mL量瓶中,加乙醇溶解并稀释至刻度,摇匀,精密量取2 mL,置5 mL量瓶中,加流动相稀释至刻度,摇匀,作为吲达帕胺对照品贮备溶液。分别精密量取杂质贮备溶液2.4 mL、杂质A贮备溶液1 mL、吲达帕胺对照品贮备溶液1 mL,置同一个10 mL量瓶中,加流动相稀释至刻度,摇匀,即得。
2.1.2.5 空白辅料溶液 取处方比例的空白辅料适量,精密称定,加乙醇适量充分振摇使溶解并稀释至刻度,摇匀,离心,取上清液2 mL加流动相稀释至5 mL,摇匀,即得。
2.1.2.6 空白溶剂 取乙醇2 mL,置5 mL量瓶中,加流动相稀释至刻度,摇匀,即得。
2.1.3 专属性实验 精密量取系统适用性溶液、空白溶剂、4-氯-3-氨磺酰胺苯甲酸对照品溶液、空白辅料溶液、供试品溶液和自身对照溶液各20 μL,注入液相色谱仪,空白溶剂及空白辅料溶液对有关物质检查无干扰,吲达帕胺与已知杂质4-氯-3-氨磺酰胺苯甲酸峰、杂质A、杂质B分离情况良好。见图1。
2.1.4 破坏性实验 取本品研碎,取细粉适量(相当于吲达帕胺15 mg),精密称定,分别通过加入5 mol·L-1盐酸溶液水浴20 min(破坏后加碱中和至中性)、加入5 mol·L-1氢氧化钠溶液室温放置14 h(破坏后加酸中和至中性)、加入30%过氧化氢溶液室温放置8 h、紫外灯[200 W·h-1·(m2)-1]下14 d 和水浴24 h 的方式进行酸性、碱性、氧化、光照和高温破坏性实验。结果见图2,吲达帕胺与各种破坏条件下产生的杂质分离情况良好,二极管阵列检测器对吲达帕胺主峰进行峰纯度分析,纯度因子均在阈值限度之内,显示主峰为单一物质。

图1 6种溶液HPLC色谱图

图2 强破坏实验HPLC图(方法一)
2.1.5 线性关系考察及校正因子的测定 取吲达帕胺、杂质B和4-氯-3-氨磺酰胺苯甲酸峰对照品各适量,精密称定,分别用流动相依次稀释制成浓度为1.790~0.030 μg·mL-1,1.005 ~ 0.038 μg·mL-1,1.178 ~0.007 μg·mL-1的6 个系列溶液,精密量取20 μL,注入色谱仪,记录峰面积。以浓度为横坐标(X),峰面积为纵坐标(Y)绘制标准曲线。以两条线性方程的斜率计算杂质B相对于吲达帕胺的校正因子f。为验证校正因子的准确性,改变实验仪器、日期、实验人员,照上述方法重复实验。由两次实验可知,杂质B相对于吲达帕胺的校正因子f为0.7。4-氯-3-氨磺酰胺苯甲酸的线性方程为Y=34 661X+242.28,r=0.999 9。
2.1.6 重复性实验 取同一批样品(批号:130101)6份,精密称定,照“2.1.2”项方法分别制成供试品溶液和自身对照溶液。在上述色谱条件下测定,供试品中均未检出4-氯-3-氨磺酰胺苯甲酸,杂质B的平均含量为0.14%,RSD 为0.81%(n=6)。
2.1.7 中间精密度实验 精密称取同一批样品(批号:130101),6份,改变分析日期、人员、设备,照“2.1.2”项下方法平行制成供试品溶液和自身对照溶液。在上述色谱条件下测定。供试品中均未检出4-氯-3-氨磺酰胺苯甲酸,结合重复性实验结果计算,杂质B 的平均含量为0.14%,RSD 为1.00%(n=12)。
2.1.8 定量限和检测限 取杂质B、吲达帕胺及4-氯-3-氨磺酰胺苯甲酸对照品各适量,精密称定,加乙醇溶解并稀释制成较浓的贮备液,再用流动相逐级稀释后进样,分别按10倍和3倍的信噪比计算其定量限和检测限。吲达帕胺、杂质B、4-氯-3-氨磺酰胺苯甲酸的检测限分别是 0.20,0.52,0.05 ng,定量限分别是0.58,1.49,0.14 ng。
2.1.9 稳定性实验 取“2.1.2”项下供试品溶液,室温放置 7 d,分别于0,2,4,6 h 和第 2,3,4,5,6,7 天时进样20 μL,记录色谱图,结果供试品中均未检出4-氯-3-氨磺酰胺苯甲酸,杂质B峰面积的RSD为1.31%,表明供试品溶液在室温放置7 d稳定。
2.1.10 加样回收率实验 取同一批样品(批号:110301)适量(约相当于吲达帕胺15 mg),9份,精密称定。分加精密加入杂质B贮备液3,6,9 mL,各浓度平行操作3份,照“2.1.2”项方法制备供试品溶液,照上述色谱条件进样测定,记录峰面积,平均回收率为100.7%,RSD=0.72%(n=9);另取4-氯-3-氨磺酰胺苯甲酸储备液,同法操作,记录峰面积,平均回收率为97.6%,RSD=1.24%(n=9)。表明本方法准确性良好。
2.1.11 外标法和加校正因子的主成分自身对照法分别测定杂质B的含量 取不同批次样品,分别采用杂质B对照品外标法和加校正因子的主成分自身对照法进行杂质B含量检查,2种测定方式的结果差异无统计学意义,见表1。
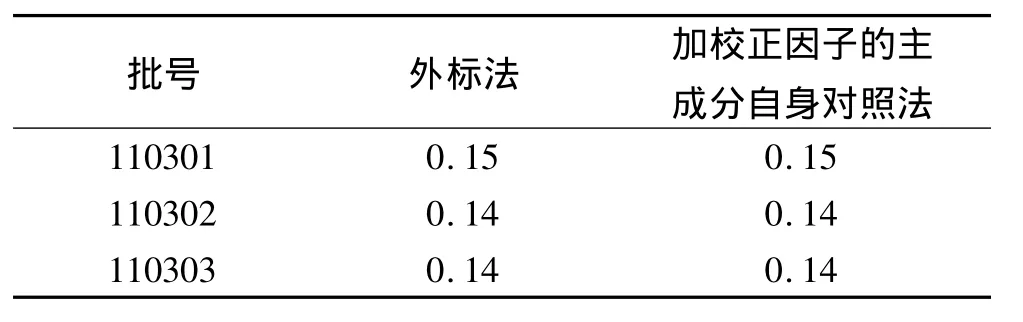
表1 吲达帕胺缓释片中杂质B测定结果(方法一) %
2.2 杂质A的测定(方法二)
2.2.1 色谱条件 Waters Symmetry Shield TM C18柱(150 mm ×4.6 mm,5 μm);以乙腈-四氢呋喃-1.5 g·L-1三乙胺溶液(用磷酸调节pH至2.8)=(7∶20∶73)为流动相;检测波长305 nm;柱温 30℃,流速1.4 mL·min-1,进样量500 μL。
2.2.2 溶液的配制
2.2.2.1 空白溶剂 取乙腈 5 mL,置 10 mL 量瓶中,加水稀释至刻度,摇匀,即得。
2.2.2.2 空白辅料溶液 称取处方比例的空白辅料适量(相当于主药量25 mg)精密称定,置25 mL量瓶中,精密加乙腈20 mL,充分摇匀,离心,精密量取上清液4 mL,置10 mL量瓶中,加水稀释至刻度,摇匀,即得。
2.2.2.3 供试品溶液 取本品细粉适量(相当于吲达帕胺25 mg),精密称定,置25 mL量瓶中,精密加入乙腈20 mL。密塞,振摇使溶解,离心,取上清液,即得供试品贮备溶液。精密量取供试品贮备溶液4 mL,置10 mL量瓶中,加乙腈1 mL,用水稀释至刻度,摇匀,即得供试品溶液。
2.2.2.4 杂质A对照品溶液 取杂质 A 对照品1.25 mg,精密称定,置于100 mL量瓶,加乙腈溶解并定量稀释制成每毫升中约含0.125 μg的溶液,即得杂质A贮备溶液。精密量取杂质A贮备溶液1 mL,置10 mL量瓶中,加水稀释至刻度,摇匀,即得杂质A对照品溶液。
2.2.2.5 混合溶液 精密量取供试品贮备溶液4 mL、杂质A贮备溶液1 mL,置同一个10 mL量瓶中,加水稀释至刻度,摇匀,即得。
2.2.3 专属性 精密量取“2.2.2”项下的空白溶剂、空白辅料溶液、供试品溶液、混合溶液、杂质A对照品溶液各500 μL,注入液相色谱仪,色谱图见图3,结果表明混合溶液中吲达帕胺峰与杂质A峰分离度良好,空白溶剂、空白辅料对样品中杂质A的检查无干扰。
2.2.4 强破坏实验 取本品细粉适量,分别通过加入5 mol·L-1盐酸溶液水浴20 min(破坏后加碱中和至中性)、加入5 mol·L-1氢氧化钠溶液室温放置14 h(破坏后加酸中和至中性)、加入30%过氧化氢溶液室温放置2 h、紫外灯[200 W·h-1·(m2)-1]下14 d和水浴24 h的方式进行酸、碱、氧化、光照和加热破坏性实验,结果显示各种破坏下产生的杂质与吲达帕胺分离良好。见图4。
2.2.5 线性关系考察 精密量取“2.2.2”项下杂质A 贮备溶液 0.6,0.8,1.0,1.2,1.6,2.0 mL,分别置10 mL量瓶中,加水稀释至刻度,摇匀,制成每毫升中含杂质 A 0.007 6 ~0.025 2 μg的系列浓度对照品溶液(相当于杂质含量 15~50 μg·g-1)。分别精密量取500 μL,注入色谱仪,记录色谱图,以浓度(X)为横坐标,峰面积(Y)为纵坐标,得线性回归方程 Y=518.03X+0.055 4,r=0.999 9。
2.2.6 稳定性实验 取“2.2.2”项下的供试品溶液和混合溶液于室温条件下放置,在0,2,4,8,24 h分别进样测定,记录色谱图,供试品溶液中均未检出杂质A,混合溶液中杂质A峰面积的RSD为1.59%,表明供试品溶液在室温放置24 h稳定。
2.2.7 精密度实验 取“2.2.2”项下的供试品溶液和混合溶液,重复进样6次,供试品溶液中均未检出杂质A,混合溶液中杂质A峰面积的RSD为1.48%。改变分析日期、人员、设备、同法测定。计算得杂质A峰面积的RSD为1.67%。

图3 空白溶剂、空白辅料、杂质A对照品、供试品和混合溶液的HPLC图

图4 强破坏实验HPLC图(方法二)
2.2.8 定量限和检测限 取“2.2.2”项下的杂质A对照品溶液,采用逐步稀释法,分别按10倍和3倍的信噪比计算其定量限和检测限。杂质A的定量限为3.0 ng,检测限为1.0 ng。
2.2.9 样品测定 取中试3批吲达帕胺缓释片(批号:110301,110302,110303),考察长期稳定性实验中杂质A的含量变化,结果见表2。

表2 吲达帕胺缓释片中杂质A含量测定结果(方法二)μg·g-1
3 讨论
本实验考察《中华人民共和国药典》2010年版(CP2010)吲达帕胺片标准[4]有关物质测定的色谱条件,流动相为甲醇-水-醋酸(45∶55∶0.1)时不能有效分离已知杂质4-氯-3-磺酰胺苯甲酸。经考察,《英国药典》2010版(BP2010)吲达帕胺片标准[5]有关物质的流动相(水-乙腈-2-丁醇-三乙胺-十二烷基硫酸钠溶液=690∶310∶20∶10∶6)能有效分离自研制剂的杂质和各种破坏条件下产生的杂质。
国内外药典标准中吲达帕胺制剂有关物质检测的供试品处理方法主要有两种,一是缓释片研细混匀后取细粉溶解,二是直接取整片溶解,振摇2 h。本品经两种方式处理后的检验结果无明显差异,鉴于本品为高分子材料骨架缓释片,取整片振摇需时长,增加杂质增长的风险。因此选择第一种处理方法。
BP2010版采用杂质对照品外标法测定杂质B含量。由于杂质B对照品昂贵且不易获得,本实验采用加校正因子的主成分自身对照法测定,方法准确可行。
BP2010版仅在吲达帕胺原料中控制杂质A的限度(5 μg·g-1),制剂中并无控制。在自研吲达帕胺缓释片稳定性研究过程中发现杂质A的含量随储存时间延长有所增加,故建立制剂中杂质A的检测方法并进行方法学验证,对其安全性进行控制。方法一中虽然能检测到杂质A,但由于自研制剂中只有微量杂质A,受方法一的进样量和检测波长(240 nm非最大吸收波长)的限制,杂质 A的内控限度上限值(50 μg·g-1)在方法一的测定条件下不能被检出,故建立方法二进一步控制杂质A的量。由于本品的杂质A属于痕量杂质,本实验采用减差法减少测定的误差。
综上所述,本实验建立的两个方法专属性强,操作简便,准确灵敏,能有效分离吲达帕胺缓释片中的杂质,能在杂质谱层面上有效控制杂质含量,保证制剂安全、质量可控。
[1]胡玉钦,于洋,杨汉煜,等.吲达帕胺缓释胶囊生物等效性及药物动力学研究[J].中国医药工业杂志,2006,37(7):483-485.
[2]谢清春,陈燕忠,吕竹芬,等.吲达帕胺缓释片的制备及体外释放度测定[J].广东药学院学报,2010,26(6):561-563.
[3]崔升淼,赵春顺,何仲贵.吲达帕胺缓释片的研制及释药机理考察[J].中国医院药学杂志,2003,23(7):397 -399.
[4]国家药典委员会.中华人民共和国药典(二部)[M].北京:中国医药科技出版社,2010:345-346.
[5]British Pharmacopoeia Commission.British Pharmacopoeia[M].London:the Stationary Office Press,2013:3020.
