植物中miRNA及其靶基因的检测与鉴定方法
2015-09-10位明明李维国
位明明++李维国


摘 要 miRNAs(microRNA,微RNA)作为植物体内一类重要的内源性非编码小分子RNA,能够显著调控细胞发育的多个生物学过程,对植物生长起重要调控作用。诸多研究表明:在植物生长发育过程中miRNA的表达具有组织特异性和时空特异性。因此,检测和分析不同组织或样品中miRNA及其靶基因的种类与表达特征,可为研究miRNA的生物学功能提供重要信息和依据。本文综述了近年来植物中miRNA及其靶基因的检测与鉴定方法及其存在的优缺点,为进一步开展miRNA及其靶基因的功能研究提供借鉴和参考。
关键词 miRNA ;靶基因 ;检测方法
分类号 Q522
Detection and Identification of miRNAs and Their Target Genes in Plants
WEI Mingming LI Weiguo
(State Centre for Rubber Breeding / Ministry of Agriculture Key Laboratory of Biology and Genetic Resources of Rubber Tree / Rubber Research Institute, CATAS,Danzhou,Hainan 571737)
Abstract miRNAs, an important class of endogenous non-coding small RNA molecules in plants, can significantly regulate multiple biological processes of cell development and play an important role in plant growth regulation. Many studies show that the expression of miRNA in the process of plant growth has spatial and temporal specificity. Therefore,detection and analysis the characterization of miRNAs and their target genes in different tissues or samples can provide important information and basis for studying the biological function of miRNA. This paper discusses the detection and identification methods of miRNAs and their target genes in plants, as well as the advantages and disadvantages of different methods in recent years,so to provide the reference and instruction for further studying the function of miRNAs and their target genes.
Keywords miRNA ; target gene ; detection methods
对于小分子RNA的关注,始于2000年6月人类基因组计划的完成,该计划的完成向人类揭示了高等哺乳动物基因组中蛋白质编码序列不足2%,而98%以上的序列都是非蛋白质编码DNA[1]。此后,随着多种模式植物基因组测序的完成,科学家发现,非编码序列占绝大多数的情况同样存在于植物基因组中。起初许多学者将此类非蛋白编码基因认为是“垃圾DNA”,但随后研究发现,生物细胞所表达的RNA中,除了前人熟悉的tRNA、rRNA、mRNA外,还蕴藏着大量不编码蛋白质的非编码RNA分子(noncoding RNA,ncRNA),这些在生物体基因组上广泛分布的非蛋白质编码区并非“垃圾DNA”,而是包含了大量转录调控元件的非编码RNA基因[2],它们能够调控真核细胞发育的多个方面,如影响功能基因的表达、调控细胞周期和个体发育等多种行为[3]。ncRNA的发现,打破了长期以来对RNA仅限于DNA的“转录信使”这一角色的认识,使科学家重新审视对细胞功能及其进化的认识。非编码RNA分子在多个物种中不断被发现和鉴定,揭示出了生物体内存在着一个巨大的且不为人知的“非编码RNA世界”。同时,基于不同物种体内非编码RNA分子的研究也拓生出了一门新的科学——RNA组学(RNomics)。目前,RNA组学研究已成为后基因组学时代的一个重要科学前沿[4],尤其是微RNA(microRNA,miRNA)的鉴定和功能研究,已成为继小干扰RNA(short interfering RNA,siRNA)之后新的研究热点之一。本研究对miRNA的验证方法、miRNA靶基因的鉴定方法进行了概述,为进一步开展植物miRNA的功能研究提供借鉴和参考。
1 miRNA研究进展
1.1 miRNA的发现
miRNA于1993年首次在秀丽新小杆线虫(caenorkabditi solegans)中被发现,当时研究人员在对线虫突变体进行遗传分析时,发现了一类长度为22个核苷酸的小分子RNA,它们能够调控细胞发育的时序,并将其命名为lin-14 RNA[5],进一步研究发现lin-14 RNA虽不编码蛋白质,却能与靶基因 lin-14的3′非翻译区(3′untranslated region,3′UTR)以碱基互补配对的方式结合,并抑制Lin-14基因蛋白的翻译,从而调控线虫的细胞发育由第一期向第二期转化。然而,当Lin-14 RNA首次被发现时,并未引起科研人员太多的关注,直到2000年另一个类似的具有细胞时序调节功能的小分子RNA——let-7 RNA被发现后,这些非编码RNA才逐步得到人们的关注[6]。由于lin-14 RNA和let-7 RNA具有显著的细胞时序调控功能,起初被命名为小时序RNA(small temporal RNA,stRNA)。随着研究的深入,在人和小鼠等哺乳动物中同样发现了大量具有转录后调节功能的小分子RNA。2001年世界不同国家的科研人员将这一类具有转录后调节功能的小分子RN再统一命名为miRNA[7-9]。
植物中miRNA于2002年首次在拟南芥小分子RNA文库中被发现。尽管植物中小分子RNA发现的时间比动物晚了将近10 a,但植物中小分子RNA的鉴定方法要比动物中完善。目前,不仅在拟南芥和水稻等模式植物中发现和验证了大量的miRNA,而且在其它植物中也发现和验证了数量可观的miRNA。随着生物体内发现的miRNA家族成员的不断增多,为了更好地对不同物种中发现的miRNA进行登记和命名,国际上建立了miRNA文库—miRBase[10-11]。截至2015年5月,在miRNA数据库 (http:microrna.sanger.ac.uk/sequenees/)中注册提交的miRNA成熟序列已达28 654条,极大地丰富了人们对生物体内非编码小分子RNA的认识。
1.2 植物miRNA的生成过程
研究发现大部分植物miRNA在基因组上具有一个独立的转录单位,只有少数miRNA在基因组上呈串联重复存在,在转录过程中由一个转录单位可加工形成多个miRNA。miRNA在植物体内的生成过程是一个复杂的、涉及多种酶和功能蛋白参与的过程[12],具体步骤如图1所示:首先植物基因组上具有特定位点的MIR基因,在RNA聚合酶的作用下转录成一个或多个较长的初级转录产物(pri-miRNA),然后初级转录物在Drosha酶的作用下[13],进一步加工形成一个具有特定二级茎环(stem-loop)结构的次级转录物(pre-miRNA),这一系列过程是在细胞核内完成的;接着pre-miRNA要在nuclear export的作用下从细胞核运输到细胞质中,由Dicer酶通过两步法进行酶切加工,随后酶切产物在HYL1(HYPONASTICLEAVES1)、HEN1(HUAENHANCER1)、HST(HASTY)等蛋白的作用下,形成成熟的miRNA[14-15]。此外,成熟的单链miRNA必须与Argonaute(AGO)蛋白相互作用形成一个RNA诱导沉默复合物(RNA-induced silencing complex,RISC),才能与目标mRNA结合而发挥作用[16-17]。
1.3 miRNA的特点
miRNA是生物体内一类5′端带磷酸基团,3′端带羟基基团的内源性小分子RNA。在生物体内miRNA是由非蛋白编码基因转录物形成的茎环结构加工而来,并以碱基互补配对的方式靶向mRNAs,进而影响目标mRNAs的翻译和稳定性。从目前的研究结果来看,miRNA主要有以下特征:(1)miRNA广泛存在于生物体内。不仅在模式植物拟南芥[18]和水稻[19]中发现了大量的miRNA,而且在一些重要农作物如玉米[20]、小麦[21]、棉花[22]、及大豆[23]中发现了大量的miRNA,甚至在苔藓、蕨类等低等植物中也发现了大量的miRNA存在[24]。(2)miRNA没有开放阅读框(ORF)及蛋白质编码基因。在动植物中绝大多数miRNA产生于基因间隔和非蛋白编码区,只有极少数miRNA产生于基因的启动子区,它们是由不同于mRNA的转录单元表达产生的[25]。(3)miRNA具有典型的长度范围,长度约为20~24 nt。大多数植物miRNA的长度都集中在21~23 nt,但miRNA的前体长度变化较大,有时长度甚至超过1 kb[26-27]。(4)不同物种中miRNA基因位点通常成簇存在。如研究发现,一些miRNA在生成过程中通常使用一个共同的前体结构,即来源于物种基因组上的同一个miRNA基因簇,并由该前体结构加工形成多个不同的成熟miRNA[28]。(5)miRNA具有典型的序列保守性。与蛋白编码基因相比,miRNA在进化上更具保守性。如在拟南芥中发现的42个miRNA家族也广泛存在于水稻、玉米和高粱等其它作物中[29]。同时,miRNA的序列保守性,也为从其它物种中鉴定并克隆miRNA提供了方便。(6)miRNA的表达具有时序性和组织特异性的特征[30-31]。miRNA在不同组织或器官中的表达量差异很大,在同一器官的不同发育时期表达量也有很大差异,表明在生物体的不同发育阶段,需要开启和关闭不同的miRNA来调控其发育进程。(7)大多数miRNA的成熟体都是从前体5′端或3′端的一条臂上加工而来,只有少数miRNA的成熟体是由其前体的两条壁上同时加工产生[32]。(8)成熟miRNA的 5′端有一个磷酸基团,3′端为羟基,且miRNA的起始碱基多为尿嘧啶核苷酸[33-34]。(9)与动物miRNA的加工和成熟过程需要Drosha、Dicer和Exportin-5等蛋白所不同的是,植物miRNA的生成过程必须经过Dicer-likel(DCL1)、Hasty(HST)、Hyponastie leaves l(HYLI),以及Hua enhancerl(HENI) 等蛋白的加工过程[35-37],这些蛋白在植物miRNA的加工和成熟过程中起着至关重要的作用,它们可直接影响植物体内miRNA的生成、种类、以及分布。(10)大多数植物miRNA与其靶基因几乎完全互补,而动物miRNA与其靶基因则不完全配对[38]。此外,植物miRNA与靶基因的结合位点不仅限于靶基因的3′非翻译区(untranslated region,UTR),还可以位于转录区域[39-40]。植物中miRNA及靶基因的高度互补性也为相关miRNA的靶基因鉴定和生物信息学预测提供了很大便利。
2 miRNA的鉴定方法
前人研究表明,在哺乳动物中miRNA的异常表达与癌症[41]、神经紊乱[42]、以及心脏疾病的发生密切相关[43-44],如在大肠癌和乳腺癌细胞中,miRNA的表达量较正常组织细胞有较大改变,人们推测miRNA可能在癌变过程中起到原癌基因和抑癌基因的作用[45]。在植物体内miRNA能够显著调控其多个生物学进程,对植物的生长发育至关重要。如与miRNA代谢及其作用机制相关的几个重要基因的突变体,均体现出发育过程中多向性的缺陷[46-47]。因此,对生物体不同组织或样本中miRNA的表达特征进行检测和分析,将有助于人们进一步了解相关miRNA与生物体发育的关系,并为相关miRNA的功能研究提供信息和依据。
由于成熟miRNA具有片段小、细胞中表达水平普遍偏低、且同一miRNA家族成员间仅有1-2个碱基的差别等特点,这些不利因素给miRNA的检测带来了较大困难和挑战。尽管如此,前人还是根据miRNA的表达特征发明了以下5种miRNA检测方法:Northern blotting法[48]、微阵列法[49]、克隆和测序法[50]、以及实时荧光定量PCR法[51]。根据miRNA的检测过程中是否需要扩增样本,又可将以上5种方法大体归为两类:一类是不需要样本扩增的探针直接杂交法,如Northern blotting法;另一类是基于样本miRNA扩增的方法,如实时荧光定量PCR法。总之,无论何种检测方法,一种理想的miRNA定量检测方法都应具有灵敏度高、特异性强、通量高和成本低等优点,才能适应生物学的发展需求。
2.1 基于探针杂交技术的Northern blotting法
基于探针杂交技术的Northern blotting法是目前最为经典的一种miRNA检测方法,该方法可以直接对样品中的miRNA进行检测,而不需要对样本中miRNA进行预扩增。它的基本原理是先将标记过的miRNA探针固定在尼龙膜上,然后再将样本RNA与经过标记的探针进行杂交,最后洗涤多余的杂交探针后进行信号检测。该方法在miRNA的检测中最为普遍,其优点是可同时对多个不同组织中的miRNA进行检测,且检测成本较低。由于经典的Northern blotting方法是基于固相探针杂交技术,所以该检测方法最大的缺点是杂交速度慢、灵敏度低;同时,该方法在检测过程需要耗费大量的RNA样本,对于那些RNA产率很低和容易降解的样品不太实用。
为了克服Northern blotting探针杂交法特异性差和灵敏度低的缺点,人们发明了用锁核酸(LNA)修饰的杂交探针来取代传统探针的方法,即改进的Northern blotting方法[52]。由于LNA是一类寡核苷酸衍生物,它对核酸分子具有较高的亲和性,可渗入到核酸序列的任何位置,因而可以提高LNA探针与目标miRNA分子结合后的双链热稳定性,使得检测的灵敏度和特异性显著提高[53]。
2.2 基于微阵列芯片的高通量检测方法
虽然传统的Northern blotting法在miRNA检测中较为常见,但该方法操作步骤繁琐,不能同时对样品中多个不同的miRNA进行检测分析,其miRNA的检测通量明显不够。为了克服这一缺点,人们又发明了miRNA微阵列芯片技术(Microarray),它可以在同一块芯片上固定多个与miRNA互补的探针,通过对样本中相关miRNA进行杂交后,最后进行相关miRNA的信号检测[54]。尽管该方法实现了miRNA的高通量检测分析,但该方法只能检测小RNA数据库中已经提交的miRNA序列,不能对样品中新的未知miRNA进行检测,导致样品中一些miRNA表达信息的缺失,这是该方法的显著缺陷。
2.3 克隆测序法
基于探针技术的Northern blotting法和微阵列芯片法都是对已知序列的miRNA进行定量检测。而克隆测序法则可以对样品中各种不同类型的miRNA进行检测分析。克隆测序法的基本原理是:首先富集样品中的小分子RNA,然后通过反转录构建样品相关的小分子RNA的cDNA文库,接着对cDNA文库进行PCR扩增,最后将PCR扩增产物连接到表达载体上进行克隆测序,并根据文库中序列出现的丰度对相关miRNA进行定量分析。由于克隆法在测序前必须对样本进行细菌克隆,然后再对每一个克隆进行测序,因而需要消耗较大的初始样本量(几百微克RNA),并投入大量的人力和财力[55]。此外,基于克隆测序的方法通常是Sanger测序法,不仅测序效率低,而且费时费工,这些都给深入研究miRNA的功能带来了诸多不便。
2.4 实时定量PCR法
前面几种方法在检测过程中都不需要对样本进行扩增,而实时定量PCR法则需要扩增样本。目前采用实时定量PCR对miRNA进行定量检测主要有以下3种方法:第一种方法称之为引物延伸定量PCR法(PE-qPCR),它的基本原理是在常规引物延伸技术(PE)的基础上,用一个加尾的特异性引物将miRNA反转录成cDNA,然后利用一个锁核酸(LNA)修饰的miRNA特异性反向引物和一个加尾的通用引物序列,对相关miRNA进行PCR扩增[56];第二种方法称之加尾法,它的基本原理是先用poly(A)聚合酶对样品总RNA进行处理,使得样品中miRNA的3′端加上poly(A)尾巴,然后对其进行反转录,使得cDNA序列加上poly(T)接头,最后再用通用引物和miRNA序列特异引物序列,进行miRNA的PCR扩增[57]。由于该方法在PCR扩增前,需要对miRNA进行加尾反应,因此该方法被称为加尾法;第三种方法称之为stem-loop RT-qPCR检测法,其原理是首先利用一条特殊的茎-环状引物将miRNA反转录成cDNA双链,再利用与miRNA互补的特异性正反向引物和探针,对目标miRNA进行定量检测。
利用以上3种方法对miRNA进行PCR扩增时,主要采用2种信号检测模式,即荧光染料法和探针法。利用荧光染料法对miRNA进行PCR扩增时,由于不能进行多重PCR反应,同时引物二聚体、单链二级结构以及错误扩增产物的出现会增加荧光值,进而影响检测结果的准确性,限制了其在miRNA定量检测中的应用;而特异性荧光定量PCR中TaqMan荧光探针法则没有上述缺点,在实际操作中被广泛采纳[58-59]。目前,从不同物种中miRNA的检测效果来看,由于荧光探针法在miRNA的PCR扩增中不仅表现出特异性强,灵敏度高,样品消耗少,定量线性范围宽等特点[60-62],能准确区分某一miRNA家族中高度同源的不同miRNA序列,甚至还能检测出只有一个碱基差异的不同miRNA表达水平,因而在miRNA的定量检测中得以广泛应用。然而,尽管探针法在miRNA的定量检测中表现出较大优势,但在实际应用过程中每一条miRNA序列都需要设计相应的探针,费用相对较高,限制了该技术的大规模应用。
2.5 高通量测序技术
植物中蕴藏着巨大数量的小分子RNA世界,而传统的Sanger测序法在测序中程序复杂,效率较低,且测序深度有限,在很大程度上制约着植物中miRNA的发现速度。为了克服传统Sanger测序法的缺点,更好的挖掘物种中未知的miRNA,自2005年科研人员发明了以大规模平行测序(Massively Parallel Signature Sequencing,MPSS)为代表的第二代高通量测序技术,来挖掘植物中蕴藏的miRNA种类[63-65]。尽管第二代高通量测序技术获得的序列较短,但它们在测序过程中可以对上万个样本同时进行测序,极大地提高了测序效率,具有速度快、成本低、覆盖度深、产出巨大等优点[66-67]。因此,非常适合小分子RNA测序,尤其是对那些拷贝数低的小分子RNA。
尤其值得一提的是,二代测序技术中Solexa测序技术较其它二代测序技术表现出了前所未有的优势。它能一次检测上亿个核苷酸片段,且测定成本仅为常规毛细管电泳测序技术成本的1%,是二代测序技术中高质量、高通量、低成本的测序法,具有很高的应用前景。该测序技术的基本流程为:先提取高质量的样品总RNA,然后用聚丙烯酰胺凝胶电泳分离大小为18~30 nt之的小RNA片段,接着用T4 RNA连接酶将该范围内的小分子RNA加上5′端接头和3′端接头,并对两端加上接头的小RNA分子进行反转录和PCR扩增。最后将PCR扩增后得到的cDNA放置在flow cell上,进行上机测序,从而直接获得所有miRNA的序列信息。
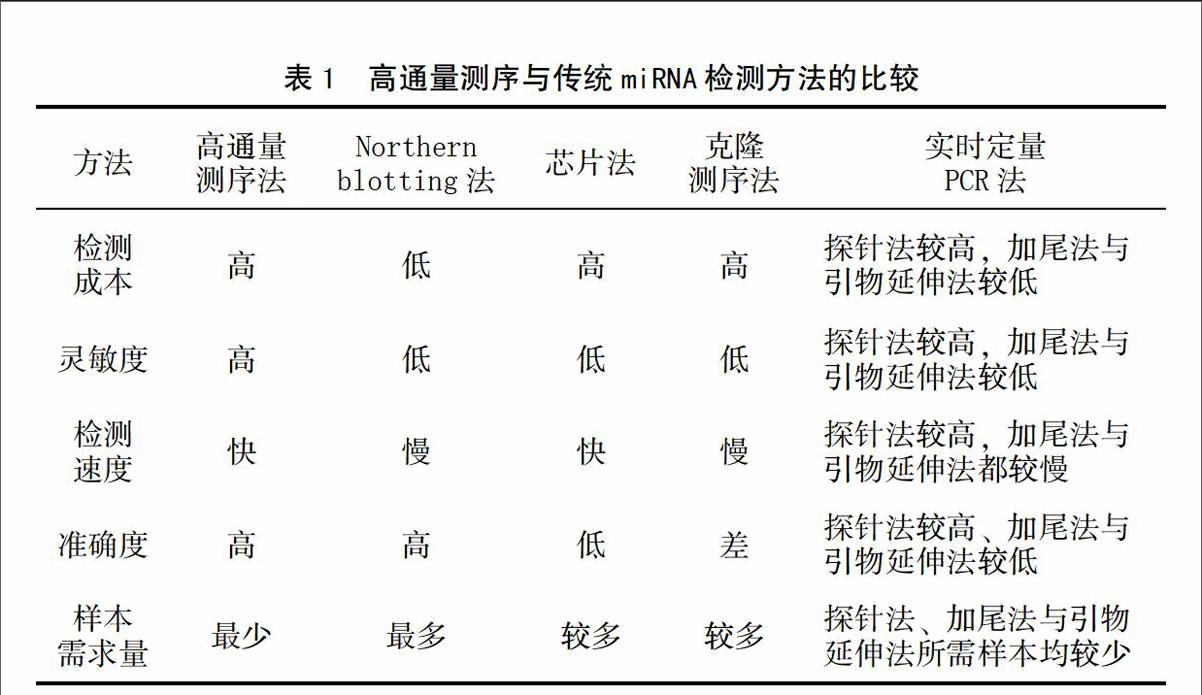
由于植物中miRNA的表达具有时空特异性,并随着诸多生物学过程的变化而发生变化,而且在植物的不同发育时期、不同生长部位其作用机理表现出较大差异,这就要求miRNA的检测过程必须具备规模化、高通量、假阳性率低等特征,而新一代高通量测序技术能很好的解决以上问题,较以往的miRNA检测方法表现出较大优势(表1)。目前,小RNA数据库中提交的绝大多数miRNA序列都是通过新一代高通量测序技术发现和鉴定而来。同时,高通量测序技术在植物学研究领域的广泛应用,不仅提升了植物中miRNA的发掘力度,促进了植物中miRNA的功能研究,提高了人们对不同物种中小分子RNA进化的认识,也为全面、深入研究植物小分子RNA的生物学功能提供最基本的素材。
3 miRNA靶基因鉴定方法
为了更好地了解miRNA的功能及其下游靶基因的相互作用关系,对相关miRNA进行靶基因鉴定就显得十分必要。目前,主要采用以下3种方法来鉴定相关miRNA的靶基因。
3.1 利用生物信息学预测miRNA的靶基因
植物体内miRNA通常与靶基因进行完全或接近完全的配对来引起mRNA的剪切,从而达到调控基因表达的目的。与动物miRNA相比,植物miRNA与靶基因的互补程度较高,这就为植物miRNA的靶基因预测带来很大便利。基于miRNA与靶基因的互补配对关系,前人发明了利用生物信息学方法来预测相关miRNA的靶基因。根据前人的研究和总结,miRNA靶基因的生物信息学预测过程中必须遵循一定的流程和规则[68-70],具体规则如下:(1)miRNA与靶基因不得超过4个碱基错配(G-U配对认为0.5个错配);(2)miRNA与靶基因的匹配过程中不能有超过2处的相邻位点碱基错配;(3)miRNA与靶基因复合体中,从miRNA的5′端起第2~12个位点不得有相邻位点发生错配;(4) miRNA与靶基因的匹配过程中不得发生第10或11位点碱基错配。大多数miRNA都在第10或第11位点对mRNA进行切割,因此这两个位点不允许有碱基错配;(5)miRNA与靶基因匹配中,从miRNA的5′端起,第1~12个位点的种子序列之间不得有超过2.5个碱基错配;(6)miRNA与靶基因匹配后的最小结合自由能(MFE),不能小于该miRNA与靶基因完全互补结合时最小接合自由能的75%。
早期miRNA的功能研究中,许多miRNA的靶基因都是通过生物信息学预测鉴定而来,当时为相关miRNA的功能研究带来了一定便利。由于传统的利用生物信息学预测miRNA靶基因的方法主要依靠计算机模拟进行验证,该方法存在一定的局限性,如相关miRNA的靶基因鉴定结果会出现数据海量,假阳性高,准确性差等弊端。因此,该方法只适合对物种间保守的miRNA进行靶基因鉴定,而对于那些未知的新miRNA进行靶基因鉴定时效果很差,这些不利因素限制了miRNA的后续研究进程。
3.2 利用5′ RLM-RACE技术鉴定miRNA的靶基因
为了从实验水平直接验证miRNA的靶基因,人们随后又发明了基于RNA连接反应的5′ Race技术来鉴定相关miRNA的靶基因[71]。该技术的原理是将提取的RNA连接上5′ Race接头,在反转录酶和GeneRacer Oligo dT 引物的作用下合成cDNA单链,然后cDNA单链在GeneRacer 5′ Primer(5′-CGACTGGAGCACGAGGACACTGA-3′)和GeneRacer 3′ Primer(5′-GCTGTCAACGATACGCTACGTAACG-3′)的作用下产生非特异性基因的5′ Race扩增产物文库。接着,使用GeneRacer 5′ Nested Primer和Gene-specific primers进行相关特异性基因的5′ Race产物扩增。最后,对特异性基因的5′ Race扩增产物进行胶回收、克隆、测序分析。
该方法虽能准确鉴定某一特定组织中相关miRNA的靶基因,但在实际应用过程中,仍存在一些不利因素[72-73]。如该技术在操作过程中需要消耗大量的样品RNA,对于那些采集比较困难的样品中miRNA的靶基因鉴定不太现实。其次,5′Race试剂盒的价格偏高,致使相关miRNA靶基因的鉴定成本过高。此外,在实际操作过程中,每次仅能从样品中鉴定有限数目的靶基因,而不能对该组织中的全部miRNA进行靶基因鉴定,使得组织样品中很多重要的靶基因切割位点信息遗失。因此,该技术只适合小范围对样品中的miRNA进行靶基因鉴定,而不能对样品中绝大多数miRNA的靶基因进行分析,这些不利因素限制了该技术在miRNA功能研究中的大规模应用。
3.3 利用降解组测序技术鉴定miRNA的靶基因
考虑到生物信息学预测miRNA靶基因时存在假阳性偏高、准确性差等弊端,而5′ Race技术又存在成本偏高,不能对组织中全部miRNA的靶基因进行高通量分析的缺点,这些不利因素困扰着miRNA的研究进程。近年来随着新一代高通量测序(High-throughput Sequencing) 技术的出现和发展,出现了一种新的称为降解组测序的方法(Degradome Sequencing) ,该技术结合了高通量测序技术与生物信息学分析的优势,能够很好的解决miRNA靶基因鉴定过程中存在的弊端和缺陷[74-76],使之在miRNA的研究中得以大规模应用。
降解组测序的原理是基于植物体内绝大多数miRNA是通过剪切作用来实现对靶基因的调控,且miRNA对靶基因的剪切位点通常发生在与mRNA互补区域的第10或第11位碱基位点上。如果mRNA受到相关miRNA的剪切作用,mRNA被剪切后就会产生两个断片——即含有帽子结构的5′端剪切片段和带有poly A尾巴的3′端剪切片段。由于mRNA被剪切后产生的3′端剪切片段含有5′单磷酸自由基团和3′poly A尾巴,该断片可被RNA连接酶连接,其连接产物可用于下一步的高通量测序;而含有帽子结构的5′端剪切片段因缺少5′单磷酸基团,无法被RNA连接酶连接,因而不能进入下一步的测序实验。紧接着使用HiSeq2000测序仪对连接产物进行测序,得到50 nt长度的原始数据。原始数据再经去接头、去污染、去低质量等步骤后获得“干净”的序列——即称为降解序列。降解序列经过一系列的注释之后,就可得到mRNA的降解片段。最后,获得的mRNA降解片段通过与miRNA的比对分析后推测出其互补序列,并借助于生物信息学工具target finder做一个反向预测来寻找mRNA-miRNA的配对关系。综合这两方面的结果,可以直观地发现在mRNA序列的某个位点会出现一个波峰,而该峰值对应的mRNA位点正是候选miRNA的剪切位点。
利用降解组测序鉴定miRNA的靶基因具有以下特点:(1)高通量性:一次测序可以检测到1 000万条以上的序列信息;(2)高准确性:从几个到几十万个拷贝的精确计数;(3)高分辨率:可以精确检测到单碱基的差异;(4)重复性好:深度测序保证了检测的随机性,不需要技术重复等特点。目前,该方法已被成功应用于水稻、棉花、玉米等重要农作物miRNA靶基因的筛选和鉴定[77-80]。同时,该方法的问世也为miRNA靶基因的鉴定和功能研究带来了革命性的变革,有利地推动了动植物miRNA的研究进程。
降解组测序技术的出现突破了传统miRNA靶基因鉴定技术的局限性,为科研人员深入开展miRNA的功能研究提供了更加灵活和有效的分析方法,该技术检测到的靶基因数量和准确率相对于生物信息学预测有较大提高,而且能大大缩小后续实验验证的范围,其实际效果是传统的研究方法所无法比拟的(表2)。在实际操作中,借助于生物学实验手段对样本中miRNA的靶基因进行高通量测序,可以快速确认上游miRNA的剪切位点及其相互作用的靶基因种类,进而得知样本中miRNA与下游靶基因的调控通径。同时,结合不同手段的生物信息学分析,如基因本体论分析(Gene Ontology,GO)、代谢通路(Pathway)分析与表型性状分析,可以从不同角度对植物miRNA的调控机制进行整体分析。目前,降解组测序技术已被广泛应用于植物生长发育调控以及逆境胁迫相关的生物学响应机制研究中,成为研究miRNA及其靶基因相互作用关系,进一步探究植物生长发育调控和胁迫相关的调节通路、以及从基因表达的转录水平和转录后水平构建调控网络的一种全新而有效的方法。
4 展望
miRNAs的发现是生命科学领域的一个重大突破,它拓展和丰富了人们对生物体内基因表达和小分子RNA调控功能的认识,促使生物学家重新反思对细胞进化的理解和认知。自1993年动物中发现第一个miRNA lin4以来,miRNA领域的相关研究得以飞速发展,并逐步成为生物学研究领域追踪的热点之一。现已证实生物体细胞中存在着数量庞大的miRNAs,它们是基因表达调控网络中丰富而重要的组成部分。在动物研究领域,诸多研究表明,miRNA与人类一些重大疾病的产生密切相关,理解miRNA介导的基因沉默机制,可对诸如癌症和心脑血管疾病产生的根本原因提供新的见解与治疗方案;在植物研究领域,miRNA与一些重要农作物产量形成的分子机制密切相关,通过对一些物种中重要农艺性状相关miRNA及其靶基因进行鉴定和功能分析,对于诠释一些重要器官中特定miRNA的生物学功能与代谢机制,破解重要农作物产量形成中的分子调控机制具有重要意义。随着miRNA及其靶基因鉴定技术的进一步完善和深入,以及大规模高通量测序技术与平台的广泛应用,必将促使人们开始对各个物种中特有的miRNA、不同发育状态下的miRNA、以及各种胁迫条件的miRNA进行挖掘和鉴定,以此来全面探究该物种中miRNA在不同条件及不同发育时期的功能。同时,随着miRNA及靶基因研究手段的进一步完善,借助于转基因技术过量表达特定的miRNA或获得靶基因缺失突变体对miRNA的功能进行验证技术的进一步完善,这些研究必将推动整个生物学研究领域的飞速发展。
纵观近年来miRNA及其靶基因检测与鉴定技术的进展,虽然鉴定并提交至小RAN数据库中的miRNA数量呈指数增加,但许多miRNA的功能仍然未知,一些提交至小RNA数据库中miRNA的真实性仍需进一步确认。在miRNA的鉴定与功能研究领域,仍存在许多未解的困惑和未知的领域。总的来说,问题主要包括以下6个方面:其一、关于物种中miRNA种类上限的问题。迄今人们不清楚任何一种物种中miRNA的种类究竟有多少,某一特定物种中发现的miRNA种类距离该物种中全部miRNA种类的上限还有多少? 其二、关于miRNA的靶基因问题。研究发现很多已经鉴定的miRNA根本找不到其作用的靶基因,或者它的靶基因功能未知。其三、关于miRNA与靶基因的接合效率和作用方式问题。在miRNA与靶基因的匹配过程中,究竟什么因素影响了miRNA与UTR位点的接近性和效率? 在什么条件下,miRNA对靶基因进行裂解或者抑制翻译? 其四、关于miRNA与靶基因的接合位点问题。研究发现很多情况下,多个不同的miRNAs会作用于同一靶基因,这些miRNA结合在同一靶基因位点上,它们如何起作用? 其五、关于miRNA靶基因功能冗余和叠加的问题。研究发现,许多miRNA具有相似的功能,它们的靶基因功能也相似。在进化中这些miRNA靶基因功能冗余和叠加的本质和意义是什么? 其六,关于miRNA与其它几种内源性小分子RNA作用机制的区别与相互联系问题。如生物体内miRNAs与siRNAs都可以导致基因表达沉默的产生,但究竟在多大程度上,miRNAs生成通路与siRNAs生产通路使用相似的同源进化基因,何种因素介导了miRNA复合体由细胞核向细胞质转运,这些问题都有待进一步研究和探讨。
参考文献
[1] International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome[J]. Nature, 2004, 31(7011): 931-945.
[2] Reinhart B J, Bartel D P. Small RNAs correspond to centromere heterochromatic repeats[J]. Science, 2002, 297(5 588): 1 831-1 831.
[3] Malone C D, Hannon G J. Small RNAs as Guardians of the Genome[J]. Cell, 2009, 136: 656-668.
[4] 屈良鹄. RNA 组学: 后基因组时代的科学前沿[J].中国科学C 辑,2009,39:1-2.
[5] Lee R C, Feinbaum R L, Ambros V, et al. Elegtms heterochronic gene lin-4 encodes small RNAs with antisense complementarity to Lin-l4[J]. Cell, 1993, 75(5): 843-854.
[6] Reinhart B J, Bartel D P. Small RNAs correspond to centromere heterochromatic repeats[J]. Science, 2002, 297(5 588): 1 831-1 831.
[7] Lagos-Quintana M, Rauhut R, Yalcin A, et al. Identification of tissue-specific microRNAs from mouse[J]. Curr Biol, 2002, 12(9): 735-739.
[8] Pasquinelli A E, Reinhart B J, Slack F, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA[J]. Nature, 2000, 408(6 808): 86-89.
[9] Lee R C, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans[J]. Science, 2001, 294(5 543): 862-864.
[10] Griffiths-Jones S, Grocock RJ, Van Dongen S, et al. miRBase: microRNA sequences, targets and gene nomenclature[J]. Nucleic acids research, 2006, 34(1): 140-144.
[11] Griffiths-Jones S, Saini H K, van Dongen S, et al. miRBase: tools for microRNA genomics[J].Nucleic acids research, 2008, 36(1): 154-158.
[12] Wierzbicki AT, Haag JR, Pikaard CS. Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes[J]. Cell, 2008, 135: 635–648.
[13] Rehwinkel J, Natalin P, Stark A, et al. Genome-wide analysis of mRNAs regulated by Drosha and Argonaute proteins in Drosophila melanogaster[J]. Mol Cell Biol, 2006, 26: 2 965-2 975.
[14] Bernstein E, Caudy A A, Hammond S M, Hannon G J. Role of a bidentate ribonuclease in the initiation step of RNA interference[J]. Nature, 2001, 409(6 818): 363-366.
[15] Jones-Rhoades M W, Bartel D P, Bartel B. MicroRNAs and their regulatory roles in plants[J]. Annu Rev Plant Biol, 2006, 57: 19-53.
[16] Hammond S C, Bernstein E, Beach D, et al. An RNA-directed nuclease mediates Post transcriptional gene silencing in Drosophila cells[J]. Nature, 2000, 404(6 775): 293-296
[17] Hutvagner G, Zamore P D. RNAi: nature abhors a double strand[J]. Curr Opin Genet Dev, 2002, 12(2): 225-232.
[18] Sunkar R, Zhu J K. Novel and stress-regulated micro-RNAs and other small RNAs from Arabidopsis[J]. Plant Cell, 2004, 16(8): 2 001-2 019.
[19] Sunkar R, Girke T, Jain PK, et al. Cloning and characterization of microRNAs from rice[J]. Plant Cell, 2005, 17(5): 1 397-1 411.
[20] Mica E, Gianfranceschi L, Pe M E. Characterization of five microRNA families in maize[J]. J Exp Bot, 2006, 57(11): 2 601-2 612.
[21] Yao Y Y, Guo G G, Ni Z Y, et al. Cloning and characterization of microRNAs from wheat (Triticum aestivum L.) [J]. Genome Biol, 2007, 8(6): 1-13.
[22] Kwak P B, Wang Q Q, Chen X S, et al. Enrichment of a set of microRNAs during the cotton fiber development[J] .BMC Genomics, 2009, 10(1): 1-11.
[23] Zhang B, Pan X, Stellwag E J. Identification of soybean microRNAs and their targets[J]. Planta, 2008, 229(1): 161-182.
[24] Arazi T, Talmor NM, Stav R, et al. Cloning and characterization of micro-RNAs from moss[J].Plant J, 2005, 43(6): 837-848.
[25] Tang G, Reinhart B J, Bartel D P, et al. A biochemical frame work for RNA silencing in plants[J]. Genes & development, 2003, 17(1): 49-63.
[26] Allen E, Xie Z, Gustafson AM, et al. microRNA-directed phasing during trans-acting siRNA biogenesis in plants[J]. Cell, 2005, 121(2): 207-221.
[27] Jung J H, Seo P J, Park C M. MicroRNA biogenesis and function in higher plants[J]. Plant Bio technol Rep, 2009, 3(2): 111-126.
[28] Sunkar R, Zhu J K. Novel and stress-regulated micro-RNAs and other small RNAs from Arabidopsis[J]. Plant Cell, 2004, 16(8): 2 001-2 019.
[29] Axtell M J, Bartel D P. Antiquity of microRNAs and their targets in land plants[J]. Plant Cell, 2005, 17(6): 1 658-1 673.
[30] Bartel D P. MicroRNAs: genomics, biogenesis, mechanism, and function[J].Cell, 2004, 116(2): 281-297.
[31] Wienholds E, Kloosterman W P, Miska E, et al. MicroRNA expression in zebrafish embryonic development[J]. Science, 2005, 309(5 732): 310-311.
[32] Ambros V. The functions of animal microRNAs[J]. Nature, 2004, 431(7 006): 350-355.
[33] Lau N C, Lim L P, Weinstein E G, et al. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans[J]. Science, 2001, 294(5 543): 858-862.
[34] Ambros V, Bartel B, Bartel DP, et al. A uniform system for microRNA annotation[J].RNA, 2003, 9(3): 277-279.
[35] Papp I, Mette MF, Aufsatz W, et al. Evidence for nuclear processing of plant micro RNA and short interfering RNA precursors[J]. Plant physi, 2003, 132(3): 1 382-1 390.
[36] Bartel B, Bartel DP. MicroRNAs: at the root of plant development[J]. Plant Physiology, 2003, 132(2): 709-717.
[37] Lund E, Güttinger S, Calado A, et al. Nuclear export of microRNA precursors[J].Science, 2004, 303(5 654): 95-98.
[38] Liave C, Xie Z, Kasschau KD, et al. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA[J].Science, 2002, 297: 2 053-2 056.
[39] Floyd S K, Bowman J L. Gene regulation: ancient microRNA target sequences in plants[J]. Nature, 2004, 428(6 982): 485-486.
[40] Wu S, Huang S, Ding J, et al. Multiple microRNAs modulate p21Cip1/Waf1 expression by directly targeting its 3′ untranslated region[J]. Oncogene, 2010, 29(15): 2 302-2 308.
[41] Cho W C S. OncomiRs: the discovery and progress of microRNAs in cancers[J]. Mol Cancer, 2007, 6(1): 60.
[42] Schetter A J, Leung S Y, Sohn J J, et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma[J]. Jama, 2008, 299(4): 425-436.
[43] Zimmerman AL, Wu S. MicroRNAs, cancer and cancer stem cells[J]. Cancer letters, 2011, 300(1): 10-19.
[44] Kong Y W, Ferland-McCollough D, Jackson TJ, et al. microRNAs in cancer management[J].The lancet oncology, 2012, 13(6): 249-258.
[45] Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer[J]. Annual review of pathology, 2014, 60: 167-179.
[46] Lu C, Fedoroff N. A mutation in the Arabidopsis HYL1 gene encoding a dsRNA binding protein affects responses to abscisic acid, auxin, and cytokinin[J]. Plant Cell, 2000, 12(12): 2 351-2 365.
[47] Schauer S E, Jacobsen S E, Meinke D W, et al. DICER-LIKE1: blind men and elephants in Arabidopsis development[J].Trends in plant science, 2002, 7(11): 487-491.
[48] Pall G S, Codony-Servat C, Byrne J, et al. Carbodiimide-mediated cross-linking of RNA to nylon membranes improves the detection of siRNA, miRNA and piRNA by northern blot[J]. Nucleic Acids Res, 2007, 35(8): 1-9.
[49] Liu C G, Calin G A, Meloon B, et al. An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues[J]. Proc Natl Acad Sci USA, 2004, 101(26): 9 740-9 744.
[50] Chen C, Ridzon D A, Broomer AJ, et al. Real-time quantification of microRNAs by stem–loop RT-PCR[J]. Nucleic acids research, 2005, 33(20): 179-187.
[51] Shi R, Chiang V L. Facile means for quantifying microRNA expression by real-time PCR[J]. Biotechniques, 2005, 39(4): 519-525
[52] Varallyay E, Burgyan J, Havelda Z. MicroRNA detection by northern blotting using locked nucleic acid probes[J]. Nat Protoc, 2008, 3(2): 190-196.
[53] Kubota K, Ohashi A, Imachi H, et al. Improved in situ hybridization efficiency with locked-nucleic-acid-incorporated DNA probes[J]. Appl Environ Microbiol, 2006, 72(8): 5 311-5 317
[54] Castoldi M, Schmidt S, Benes V, et al. miChip: an array-based method for microRNA expression profiling using locked nucleic acid capture probes[J]. Nat Protoc, 2008, 3(2): 321-329.
[55] Lagos-Quintana M, Rauhut R, Meyer J, et al. New microRNAs from mouse and human[J]. Rna, 2003, 9(2): 175-179.
[56] Allawi H T, Dahlberg J E, Olson S, et al. Quantitation of microRNAs using a modified Invader assay[J]. Rna, 2004, 10(7): 1 153-1 161.
[57] Raymond C K, Roberts B S, Garrett-Engele P, et al. Simple, quantitative primer-extension PCR assay for direct monitoring of microRNAs and short-interfering RNAs[J].RNA, 2005, 11(11): 1 737-1 744.
[58] Chen C, Ridzon D A, Broomer A J, et al. Real-time quantification of microRNAs by stem-loop RT-PCR[J]. Nucleic acids research, 2005, 33(20): 179-179.
[59] Varkonyi-Gasic E, Wu R, Wood M, et al. Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs[J]. Plant methods, 2007, 3(1): 1-12.
[60] Kramer M F. Stem-Loop RT-qPCR for miRNAs[J]. Current protocols in molecular biology, 2011, 15(10): 1-15.
[61] Hurley J, Roberts D, Bond A, et al. Next-generation microRNA expression profiling technology[J]. Humana Press, 2012: 33-52.
[62] Salone V, Rederstorff M. Stem-Loop RT-PCR Based Quantification of Small Non-Coding RNAs[J]. Small Non-Coding RNAs: Methods and Protocols, 2015, 8(10): 103-108.
[63] Brennecke J, Aravin A, Stark A, et al. Discrete Small RNA-generating loci as master regulators of transposon activity in Drosophila[J]. Cell, 2007, 128(6): 1 089-1 103.
[64] Margulies M, Egholm M, Altman WE, et al. Genome sequencing in microfabricated high-density picolitre reactors[J]. Nature, 2005, 437(7 057): 376-380.
[65] Cokus S J, Feng S, Zhang X, et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning[J]. Nature, 2008, 452(7 184): 215-219.
[66] Sánchez-León N, Arteaga-Vázquez M, Alvarez-Mejía C, et al. Transcriptional analysis of the Arabidopsis ovule by massively parallel signature sequencing[J]. J Exp Bot, 2012, 63(10): 3 829-3 842.
[67] Nuttle X, Itsara A, Shendure J, et al. Resolving genomic disorder-associated breakpoints within segmental DNA duplications using massively parallel sequencing[J].Nature protocols, 2014, 9(6): 1 496-1 513.
[68] Qiu CX, Xie FL, Zhu YY, et al. Computational identification of microRNAs and their targets in Gossypium hirsutum expressed sequence tags[J]. Gene, 2007, 395: 49-61.
[69] Rhoades MW, Reinhart BJ, Lim LP, et al. Prediction of plant microRNA targets[J]. Cell, 2002, 110: 513-520.
[70] Meyers BC, Axtell MJ, Bartel B, et al. Criteria for annotation of plant microRNAs[J]. Plant Cell, 2008, 20(12): 3 186-3 190.
[71] Liave C, Xie Z, Kasschau KD, et al. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA.Science, 2002, 297: 2 053-2 056.
[72] Wang C, Han J, Korir NK, et al. Characterization of target mRNAs for grapevine microRNAs with an integrated strategy of modified RLM-RACE, newly developed PPM-RACE and qPCRs[J]. Journal of plant physiology, 2013, 170(10): 943-957.
[73] Shangguan L, Song C, Han J, et al. Characterization of regulatory mechanism of Poncirus trifoliata microRNAs on their target genes with an integrated strategy of newly developed PPM-RACE and RLM-RACE[J]. Gene, 2014, 535(1): 42-52.
(下转第36页)
