混合丁烯/异丁烷硫酸烷基化反应动力学
2015-08-22李迪孙伟振奚桢浩张明华赵玲
李迪,孙伟振,奚桢浩,张明华,赵玲
(华东理工大学化学工程联合国家重点实验室,上海 200237)
引 言
异丁烷与低分子烯烃(乙烯、丙烯或丁烯)在强酸性催化剂条件下反应生成含有C6、C7或C8组分烷基化油的烷基化反应是炼油厂重要反应过程之一[1]。自1938年起,以浓硫酸和氢氟酸作为催化剂的异丁烷与丁烯烷基化反应工艺已实现工业应用[2]。这类烷基化油因具有高辛烷值、低敏感性、低蒸气压、燃烧清洁等突出优点受到了越来越多的重视和需求[3]。特别是烷基化油不含硫、氮组分,可显著减少车辆发动机NOx的排放,后者为大气PM2.5二次生成的主要来源。
烷基化过程传统工艺以浓硫酸和氢氟酸作为催化剂,近年来离子液体和固体酸催化剂具有绿色、环境友好等优点受到广泛关注,极有潜力成为未来烷基化发展的主流方向[4-7]。由中国石油大学重质油国家重点实验室开发的年产12万吨离子液体催化碳四烷基化工业装置已于2013年建成投产,是世界首套离子液体烷基化工业生产装置。尽管如此,传统的硫酸法烷基化流程经济性好、可靠性高等特点使其在未来相当长一段时期内仍将为烷基化油生产的主导工艺。众所周知,研究反应动力学对于设计和优化工业反应过程以及开发新型反应器都具有重要意义[8-9]。Lee等[10]结合质量传递和化学反应得到了硫酸烷基化动力学简化模型及速率常数,Langley等[11]对异丁烷/丙烯硫酸烷基化动力学进行了研究,韩明汉等[12]研究了苯与丙烯动力学。Sun等[13]考虑烷基化油关键组分建立了基于正碳离子反应机理的异丁烷/异丁烯烷基化动力学模型。然而,有关不同丁烯异构体对烷基化反应影响规律的研究国内外鲜有报道。国内炼油厂C4烯烃原料的差异以及市场对烷基化汽油的需求使得该问题的研究具有现实性和紧迫性。
本研究中,采用间歇反应装置和硫酸催化剂,在276.2 ~285.2 K反应温度范围内研究了混合丁烯/异丁烷烷基化反应动力学。实验考察了不同温度和反应时间4类关键异构烷烃异辛烷(isooctane)、三甲基戊烷(TMPs)、二甲基己烷(DMHs)、高碳(HEs)的组分分布,及TMPs/DMHs和烷基化产物总体辛烷值。并以碳正离子反应机理建立的烷基化动力学模型为基础,通过对3类关键异构烷烃组分TMPs、DMHs、HEs实验值的拟合回归获得关键模型参数,为烷基化工业反应器的设计优化提供参考。
1 实验部分
1.1 试剂原料
硫酸,分析纯(95%~98%),上海凌峰化学试剂有限公司;混合烃,异丁烷/1-丁烯/顺-2-丁烯/ 反-2-丁烯/异丁烯(87.1%/2.39%/5.65%/3.44%/1.42%,质量分数),上海神开标准气体有限公司;无水乙醇,分析纯(99.5%),国药集团化学试剂有限公司;四氯化碳,分析纯(99.5%),上海凌峰化学试剂有限公司;高纯氮气(99.9%),上海南耀气体公司。
1.2 实验装置与过程
烷基化间歇装置如图1所示。实验过程:向干燥反应釜内注入270 ml浓硫酸,通入氮气吹扫反应装置2~3次,排除系统中残留的空气和水蒸气,然后通入氮气加压至0.5 MPa,开启循环冷却装置和进料冷却装置,冷却反应釜中硫酸至设定温度,打开烃进料阀门,用高压计量泵迅速注入180 ml烃类混合原料,开启搅拌(搅拌转速为3000 r·min-1;研究表明,当转速>2500 r·min-1时传质影响基本消除[13])并开始反应计时,反应过程中,调节循环冷却装置开关控制釜内反应温度稳定在设定范围,在确定的时间点处取样,取出的液相样品经静置、称量、萃取、分液等操作后进行色谱分析。
1.3 分析方法
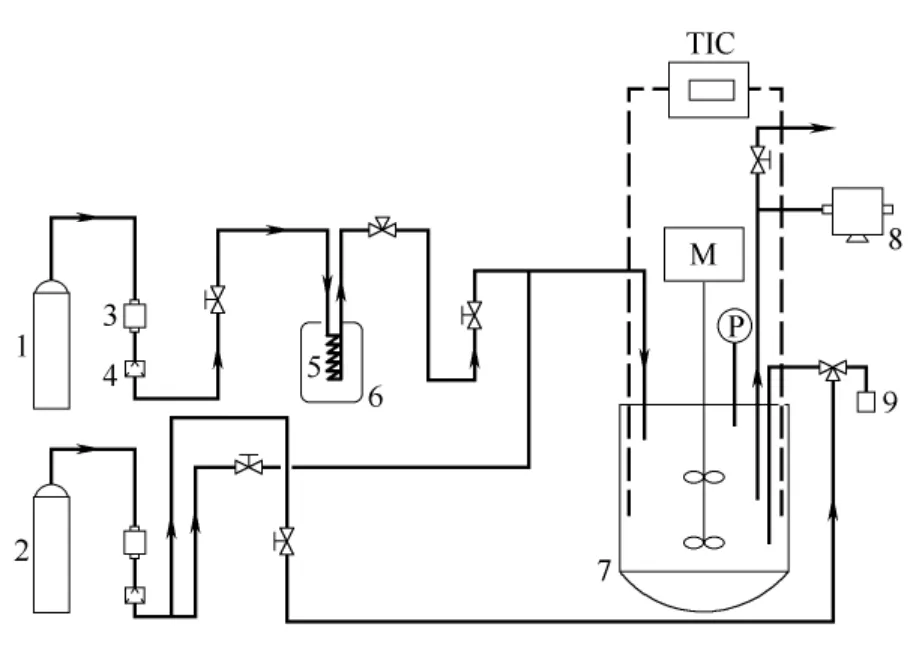
图1 间歇反应实验装置 Fig.1 Flow diagram of batch experiment setup
烷基化产物的分析采用气相色谱的方法。气相样品组分分析:天美GC7890F,色谱柱HP-AL/S,FID检测器,检测器温度180℃;载气高纯氮,进样口温度100℃。升温程序:65℃保温15 min,然后以2℃·min-1升温至160℃,继续以30℃·min-1速率升温至250℃,保温10 min。液相烷基化油组分分析:Agilent Technology GC7890A,毛细管色谱柱。升温程序:60℃柱温保持3 min,后以5℃·min-1速率升温至100℃,继续以10℃·min-1的速率升温至180℃,保持3 min。
液体烷基化油的典型GC谱图如图2所示。由于烷基化主要产物相对于异辛烷的校正因子在0.98~1.02之间,达到了期望的FID检测器上与物质苯相似的标准,因此本文采用面积归一法进行色谱定量。

图2 烷基化产物气相色谱图 Fig.2 Chromatogram of all alkylate components
2 实验结果与讨论
2.1 烷基化反应动力学的温度效应
工业反应条件下分别测定了276.2、279.2、282.2、285.2 K 4个反应温度混合丁烯烷基化动力学数据。鉴于烷基化反应速度很快,为消除传质的限制,实验中采用的搅拌转速为3000 r·min-1。同时,酸烃比1.5:1、反应体积450 ml、反应时间20 min等实验条件均保持一致。
从图3和图4可见,温度和反应时间对于异辛烷(isooctane)及三甲基戊烷(TMPs)选择性有着明显影响,在相同条件下均随着温度的降低和反应时间的延长逐渐增加。反应前5 min异辛烷和三甲基戊烷的增幅较为显著,随后增幅减缓并趋于稳定,20 min时异辛烷含量占三甲基戊烷总量约43%。反应温度由285.2 K下降至276.2 K,20 min时异辛烷及三甲基戊烷的量分别提升约10.3%和14.4%。

图3 温度和反应时间对异辛烷的影响 Fig.3 Effect of temperature and reaction time on isooctane
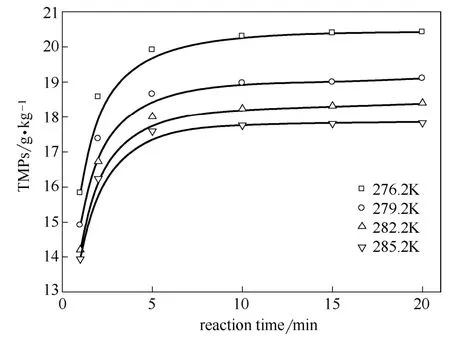
图4 温度和反应时间对三甲基戊烷的影响 Fig.4 Effect of temperature and reaction time on TMPs
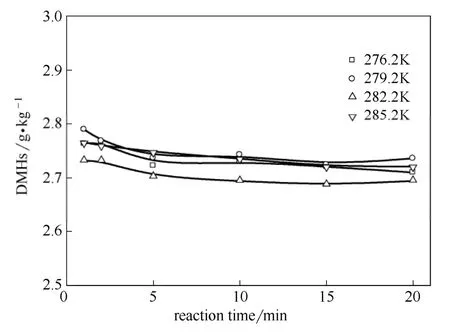
图5 温度和反应时间对二甲基己烷的影响 Fig.5 Effect of temperature and reaction time on DMHs
如图5所示,温度和反应时间对于二甲基己烷(DMHs)的影响较小,随着反应时间的延长,DMHs有着略微降低的趋势。如图6所示,TMPs/DMHs随着温度的降低和反应时间的延长而增大,总体稳定在6~8的范围,与工业反应结果一致。
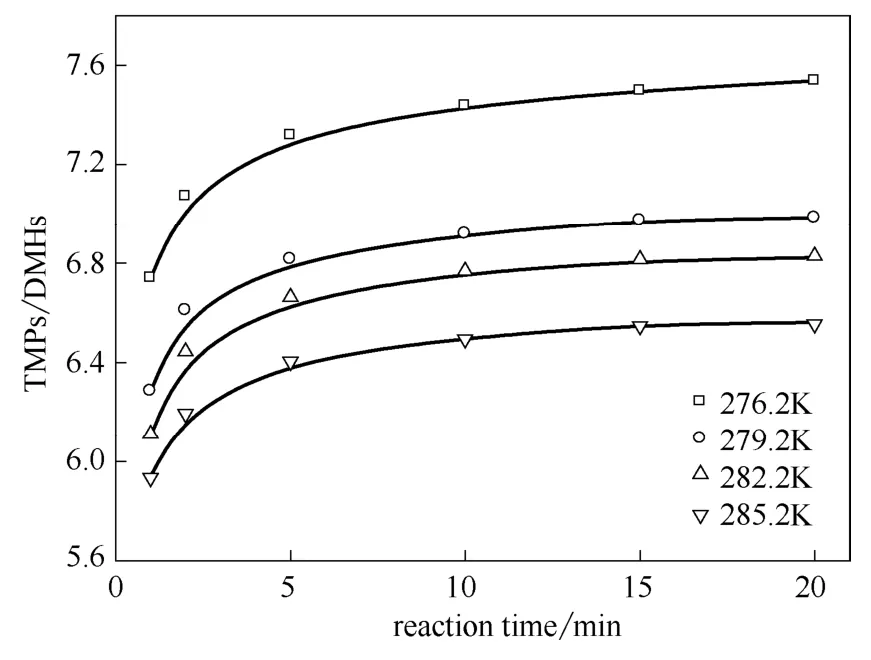
图6 温度和反应时间对TMPs/DMHs的影响 Fig.6 Effect of temperature and reaction time on TMPs/DMHs
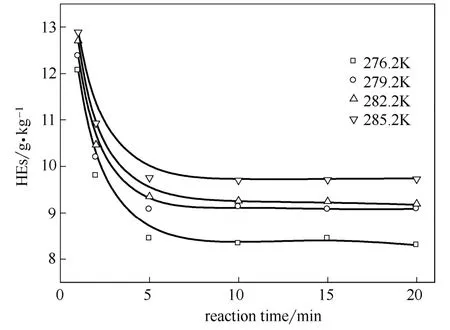
图7 温度和反应时间对高碳的影响 Fig.7 Effect of temperature and reaction time on HEs
从图7可以看出,温度和反应时间对于高碳组 分(HEs)有着较为显著的影响,同TMPs相反,反应5 min前HEs降幅明显,随后降幅减缓并趋于稳定。同时,降低反应温度生成的HEs组分明显减少,由此说明低温对于副反应有着明显的抑制作用,这对于提升烷基化产品质量有着积极的意义。如图8所示,反应初期辛烷值不断提升,5 min之后辛烷值已基本稳定,276.2 K下20 min时的烷基化产物辛烷值达到92.7。降低温度会明显提升辛烷值水平,这主要是源于低温促进了TMPs产生同时抑制了HEs的生成。
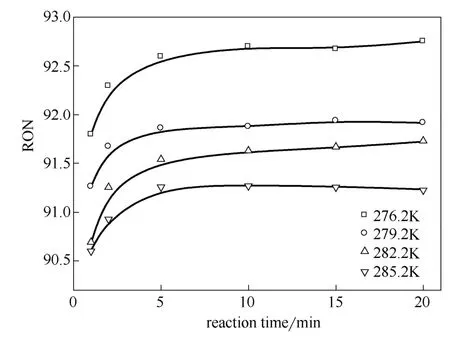
图8 温度和反应时间对辛烷值的影响 Fig.8 Effect of temperature and reaction time on RON
综合实验结果可知:在混合丁烯/异丁烷烷基化反应中,低温及较长的反应时间对于目标产物TMPs选择性及烷基化油质量有利,当反应时间大于10 min之后影响不大;烷基化产物分布和烷基化油质量对反应温度敏感,在3~12℃范围内变化较大,因此在工业操作过程中需要控制反应温度变化幅度。
2.2 烷基化反应动力学模型
目前多数研究认为,酸性催化剂上异构烷烃与C3~C5烯烃上的烷基化反应遵循以碳正离子反应机理为基础的链式反应机理,主要分为链引发、链增长和链终止3个过程。此机理最初是由Schmerling[14-15]提出的以Whitmore[16]碳正离子为基础的链式反应机理,Albright等[17-18]、Kramer[19]、Hofmann等[20-21]对此也进行了研究,趋于更加具体、成熟的碳正离子-链式反应机理普遍被人们所接受,这也为理解和研究烷基化反应过程,建立动力学模型提供了基础。通过对此机理的理解,可对烷基化反应步骤进行描述,详细的硫酸烷基化反应路线如图9所示,动力学方程及回归的动力学参数可见文献[13],其中的动力学模型微分方程如下所示。
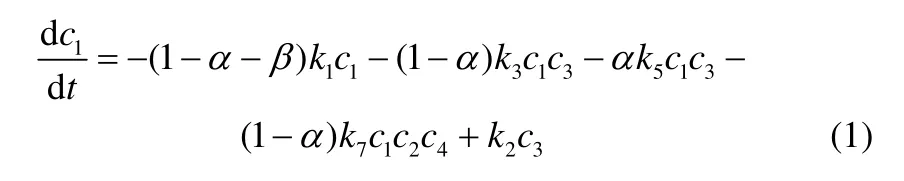

图9 硫酸烷基化反应网络 Fig.9 Diagram of reaction pathways network
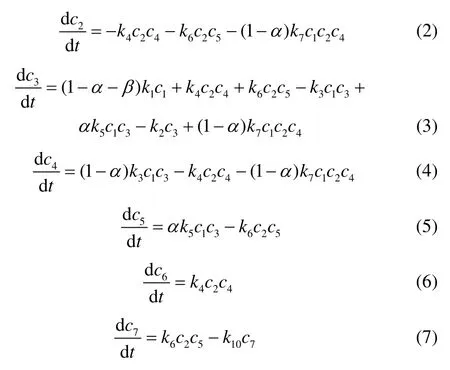
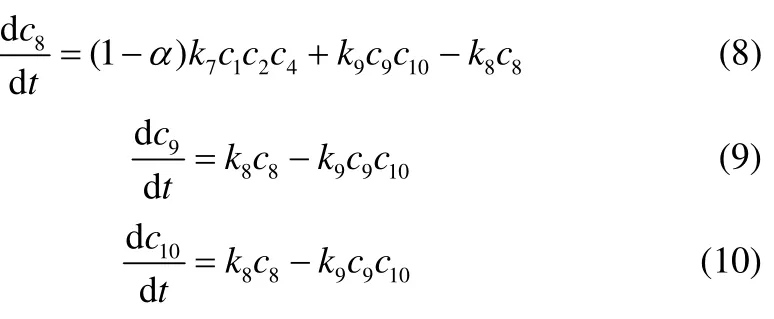
式中,下角标1为butane,2为 i- C4,3为 i-,4为TMP+,5为DMH+,6为TMP,7为DMH,8为HE,9为 i-,10为i-。
此工作中采用原料为混合烯烃,假定烷基化开始前其完全异构化,根据热力学平衡计算得到烯烃组分分布。此外,沿用了文献[13]中的模型简化方法,认为部分涉及链传递和链终止步骤反应活化能很小,假定在实验温度范围内的这些反应速率常数不变。本文保持部分速率常数与文献[13]一致,即k4及 k6~k10(1.03 kg·mol-1·min-1,4.36 kg·mol-1·min-1,88.46 kg·mol-2·min-2,0.63 min-1,5.48 kg·mol-1·min-1,2.61×10-2min-1)。在计算链引发步骤的碳正离子生成速率常数k1及其逆反应速率常数k2的同时,增加生成TMPs+和DMPs+两个反应步骤速率常数(即k3和k5)的计算。
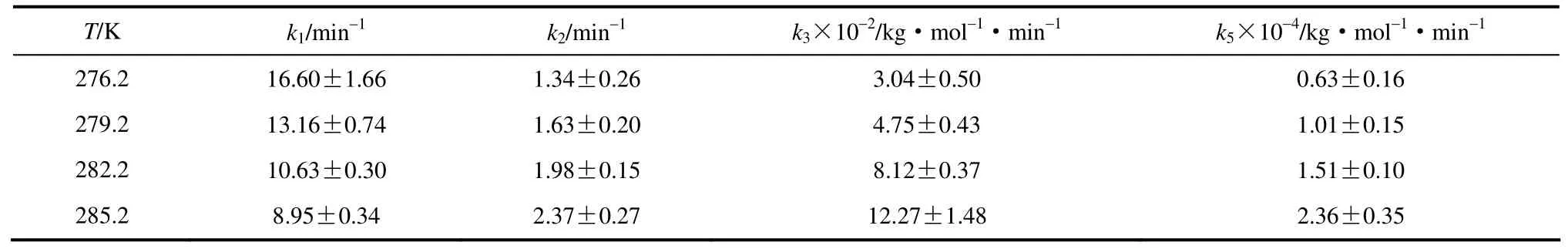
表1 速率常数估计及95%置信区间 Table l Estimated rate constants with 95% confidence intervals
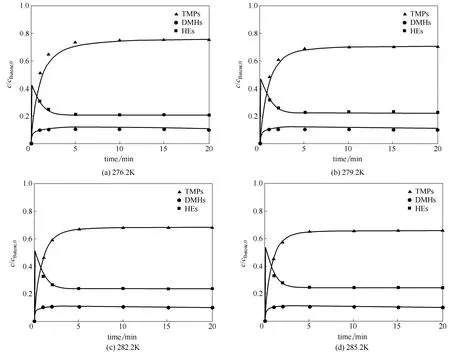
图10 烷基化反应三组关键产物的浓度分布 Fig.10 Concentration profile of key components in alkylation
采用最小二乘法对参数进行估算,速率常数估计及95%的置信区间如表1所示,置信区间比相应的参量小1~2个数量级,表明参数估计的误差较小。同时,对高碳和二甲基己烷的裂解速率即k9和k10进行微调,即k9=(4.52±0.20) kg·mol-1·min-1,k10=(7.86×10-3±3.17×10-3) min-1,可明显改善计算值与实验值的吻合结果,如图10所示。与文献[13]中相同,对于k1可发现一种反Arrhenius行为,即随着温度的升高k1值下降。值得指出的是:此现象是自由基分子存在下某些烃类反应的正常现象,是由于减少了多步反应的平衡常数的结果,在其他烃类氧化反应中也存在[22]。通过绘制lnki-1/T图获得k1、k2、k3、k5与温度的Arrhenius关系,如图11所示,线性关系良好。计算获得4个反应活化能依次为45.14、41.44、103.44、95.01 kJ·mol-1,相对应的指前因子分别是4.78×10-8min-1、9.23×107min-1、1.10×1022kg·mol-1·min-1、5.92×1021kg·mol-1·min-1。
3 结 论
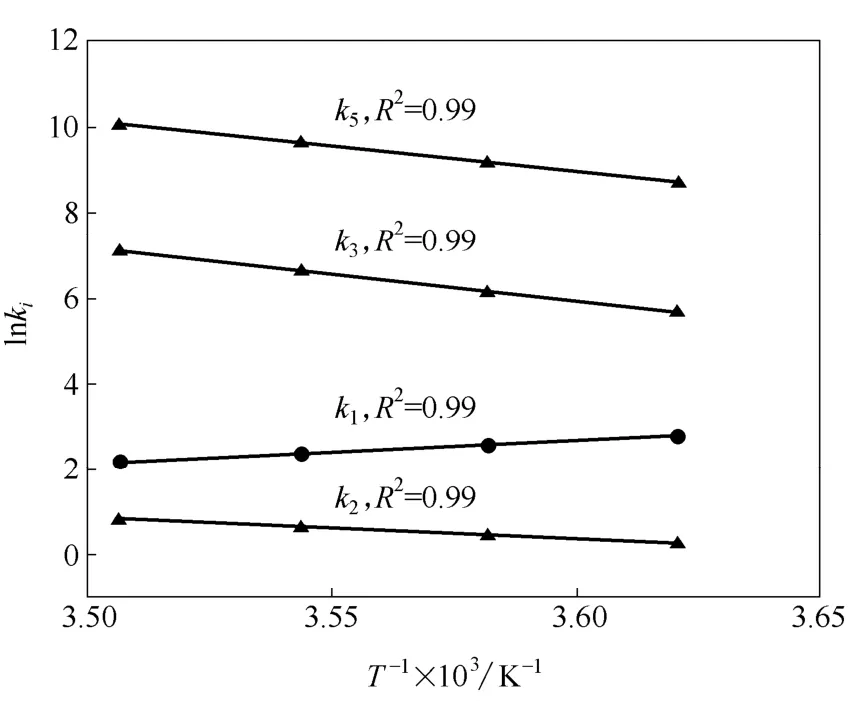
图11 lnki与T-1间的Arrhenius关系 Fig.11 Arrhenius relationship between lnkiand T-1
(1)通过间歇实验测定了276.2、279.2、282.2、 285.2 K 4个反应温度下混合丁烯/异丁烷硫酸烷基化反应动力学数据,结果表明:降低反应温度有利于烷基化主产物三甲基戊烷(TMPs)生成,而副产物二甲基己烷(DMHs)影响较小,但高碳组分(HEs)的生成量降低明显,辛烷值得到提升,因此降低温度对提升烷基化油质量有积极意义。
(2)采用基于碳正离子反应机理建立的烷基化动力学模型对TMPs、DMHs、HEs 3类烷基化关键组分进行计算分析,模型计算值与动力学实验数据符合结果良好;该动力学模型及其简化方法具有合理性,可为烷基化工业反应器的设计优化提供 借鉴。
[1] Corma A, Martínez A.Chemistry, catalysts and processes for isoparaffin-olefin alkylation: actual situation and future trends [J].Catalysis Reviews - Science and Engineering, 1993, 35 (4): 483-570
[2] Geng Yingjie (耿英杰).Industrial Alkylation and Technology (烷基化生产工艺与技术) [M].Beijing: China Petrochemical Press, 1993: 9-11
[3] Bi Jianguo (毕建国).Advances in alkylate manufacture technology [J].Chemical Industry and Engineering Process(化工进展), 2007, 26 (7): 934-939
[4] Peng Pu (彭朴), Xi Lei (葸雷), Ma Chuanyan (马传彦), Duan Qiwei (段启伟).Research process of chloroaluminate ionic liquids used as alkylation catalyst and prospect commercial application [J].Chemical Industry and Engineering Process(化工进展), 2007, 26 (4): 456-458
[5] Liu Ying (刘鹰), Liu Zhichang (刘植昌), Xu Chunming (徐春明), Zhang Rui (张睿).Study on room temperature ionic liquid-catalyzed alkylation of isobutane with butene at pilot-plant scale [J].Chemical Industry and Engineering Process(化工进展), 2005, 24 (6): 656-660
[6] Liu Ying (刘鹰), Liu Zhichang (刘植昌), Xu Chunming (徐春明).Alkylation of isobutane and 2-butene in inhibited chloroaluminate ionic liquids [J].Journal of Chemical Industry and Engineering (China) (化工学报), 2005, 56 (11): 2119-2123
[7] Zheng Dongmei (郑冬梅).Advances in alkylation process of isobutane with butene technology [J].Chemical Industry and Engineering Process(化工进展), 2004, 23 (z1): 58-61
[8] Sun Weizhen (孙伟振), Huang Huan (黄欢), Gu Xiaowu (顾晓吴), Zhao Ling (赵玲).Modeling of liquid phase oxidation kinetics of aromatic hydrocarbon based on free radical chain reaction mechanism [J].CIESCJournal (化工学报), 2010, 61 (7): 2119-2123
[9] He Zhiyong (何志勇), Luo Jun (罗军), Lü Chunxu (吕春旭), Xu Rong (徐容), Li Jinshan (李金山).Nitrolysis kinetics of DADN by N2O5/HNO3[J].CIESCJournal (化工学报), 2013, 64 (4): 1270
[10] Lee Lien-mow, Harriott P.The kinetics of isobutane alkylation in sulfuric acid [J].Industrial & Engineering Chemistry Process Design and Development, 1977, 16 (3): 282-287
[11] Langley J R, Pike R W.The kinetics of alkylation of isobutene with propylene [J].American Institute of Chemical Engineers Journal, 1972, 18 (4): 698-705
[12] Han Minghan (韩明汉), Li Xiaojin (李晓锦), Lin Shixiong (林世雄).Alkylation of benzene with propylene over βzeolite chemical reaction kinetics [J].Journal of Chemical Industry and Engineering (China) (化工学报), 1999, 50 (1):65-69
[13] Sun Weizhen, Shi Yi, Chen Jie, Xi Zhenhao, Zhao Ling.Alkylation kinetics of isobutane by C4olefins using sulfuric acid as catalyst [J].Industrial & Engineering Chemistry Research, 2013, 52 (44): 15262-15269
[14] Schmerling L.The mechanism of the alkylation of paraffins [J].Journal of the American Chemical Society, 1945, 67: 1778-1783
[15] Schemerling L.Reaction of hydrocarbons [J].Industrial & Engineering Chemistry Research, 1953, 45 (7): 1447-1455
[16] Whitmore F C.The common basis of intra-molecular rearrangements [J].Journal of the American Chemical Society, 1932, 54 (8): 3274
[17] Albright L F, Spalding M A, James A.Alkylation of isobutane with C4olefins (Ⅰ): First-step reactions using sulfuric acid catalyst [J].Industrial & Engineering Chemistry Research, 1988, 27: 381-386
[18] Albright L F.Mechanism for alkylation of isobutane with light olefins//Industrial and Laboratory Alkylations[C].Albright L F, Goldsby A R, Eds.ACS: Washington, D C, 1977, 55: 128-146
[19] Kramer G M.Hydride transfer reactions in concentrated sulfuric acid [J].Journal of Organic Chemistry, 1965, 30: 2671
[20] Hofmann J E, Schriesheim A J.Ionic reactions occurring during sulfuric acid catalyzed alkylation (Ⅱ): Alkylation of isobutane with C14-labeled butenes [J].Journal of the American Chemical Society, 1962, 84: 957-961
[21] Hofmann J E.Ionic reactions occurring in sulfuric acid (Ⅲ): The sulfuric acid catalyzed self-alkylation of isobutane with C3—C6olefins [J].Journal of Organic Chemistry, 1964, 29: 1497
[22] Lebedeva N V, Nese A, Sun F C, Matyjaszewski K, Sheiko S S.Anti-Arrhenius cleavage of covalent bonds in bottlebrush macromolecules on substrate [J].Proceedings of the National Academy of Sciences of the United States of America, 2012, 109 (24): 9276-9280
