Zn(OAc)2/AC催化固定床上CO2与1,2-丙二醇合成 碳酸丙烯酯及其反应机理
2015-08-22张光洁耿艳楼安华良赵新强王延吉
张光洁,耿艳楼,安华良,赵新强,王延吉
(河北工业大学化工学院,绿色化工与高效节能河北省重点实验室,天津 300130)
引 言
碳酸丙烯酯(PC)是一种性能优良的低毒、高沸点有机溶剂和有机合成中间体,广泛应用于有机合成、气体分离以及电化学等领域。酯交换法合成碳酸二甲酯(DMC)反应的工业化开辟了PC应用的新领域,但该工艺存在副产大量1,2-丙二醇(PG)和原料PC来源受石化工业限制两方面问题。CO2作为一种主要的温室气体,近年来其有效利用研究成为世界性课题。以CO2和PG为原料合成PC,不仅可以将酯交换法合成DMC的副产物PG重新转化为其原料PC,还可以有效利用温室气体CO2,且该反应副产物只有H2O,对于资源综合利用和保护生态环境都有非常积极的意义。
Tomishige等[1-2]报道了以CeO2-ZrO2为催化剂、乙腈为溶剂,利用PG与CO2直接合成PC的新反应路线,PC的选择性超过99%,但其收率仅为2.0%。Du等[3]以二丁基氧化锡和二丁基二甲氧基锡为催化剂、N,N-二甲基甲酰胺为溶剂对该反应进行了研究,PC收率仍然较低,最高只有3.4%。同时他们提出了二丁基氧化锡的催化机理:二丁基氧化锡首先活化PG形成2,2-二丁基-5-甲基-1,3-二氧戊环,CO2容易插入SnO键形成环状锡碳酸酯,该环状锡碳酸酯羰基碳原子上的烷氧基基团容易发生分子内亲核取代,在脱掉PC的同时催化剂得到还原。但有机锡类催化剂存在毒性大且难以回收等缺点。随后,他们开发了毒性较低的Mg和MgO代替有机锡用于催化CO2与PG合成PC反应[4],PC的收率与以有机锡为催化剂时相比略低,但其选择性最高可达100%。赵新强等[5-7]先后评价了K2CO3、K2CO3/AC和Zn(OAc)2在CO2与PG合成PC反应中的催化性能,PC的选择性分别为53.0%、74.4%和64.1%,收率分别为12.6%、9.6%和12.3%。黄世勇等[8-11]研究了1, 5, 7 -三氮杂双环[4, 4, 0]癸-5-烯(TBD)、FeCl3和KI/ZnO对以PG和CO2为原料合成PC反应的催化效果,PC的收率分别为22.5%、26.5%和26.0%。他们还推测了TBD催化CO2与PG合成PC的反应机理:在乙腈溶剂中,TBD与PG分子间通过NHO键结合成新的化合物,该化合物容易与CO2反应生成PC,在生成PC的同时催化剂TBD得到还原。上述研究都是在高压反应釜中进行PC合成反应,尚未见到关于在连续反应装置(如固定床)中进行该反应的文献报道。
有鉴于此,在本课题组已确定在高压反应釜上Zn(OAc)2对CO2与PG合成PC反应有较好催化性能的基础上[7],本文制备了负载型Zn(OAc)2催化剂,尝试用于在固定床上催化CO2与PG合成PC反应,以期探讨在连续反应装置上进行该反应的可能性。首先,确定了制备负载型Zn(OAc)2催化剂的载体和负载量,然后,考察了反应条件对PC合成反应的影响。在此基础上,采用原位红外与设计实验相结合的方法分别对CO2和PG与无水乙酸锌之间的作用情况进行了分析,并推测了无水乙酸锌的催化作用机理。
1 实验部分
1.1 主要原料及化学试剂
1,2-丙二醇,分析纯,天津化学试剂有限公司;CO2,≥99.9%,天津市四知气体有限公司;碳酸丙烯酯,分析纯,天津市化学试剂研究所;乙酸锌,分析纯,天津市江天化工技术有限公司;乙腈,分析纯,天津市康科德科技有限公司。
1.2 催化剂制备
负载型乙酸锌催化剂采用等体积浸渍法制备。以制备负载量为40%(质量分数)的Zn(OAc)2/AC催化剂为例:将活性炭破碎至0.38~0.83 mm,于烘箱中150℃干燥4 h;称取1.436 g Zn(CH3COO)2·2H2O配成5.1 ml水溶液,将其浸渍于3.0 g 活性炭上;室温老化12 h后,于旋转蒸发器中,在真空度0.07 MPa和温度70℃下处理2 h除水;在管式炉中氮气保护条件下对催化剂进行热处理[12](160℃,4 h),从而制得Zn(OAc)2/AC催化剂。
1.3 催化剂表征
采用日本理学D/MAX-2500型X射线衍射仪对催化剂样品进行物相分析。操作条件为:Cu靶,石墨单色滤光片,狭缝SS/DS=1°,RS 0.15 mm,工作电压为40 kV,电流100 mA,扫描范围10°~80°,扫描速率8 (°)·min-1。
催化剂样品的比表面积及孔结构测定在美国Micromeritics公司生产的 ASAP2020M+C型比表面积及孔隙度分析仪上进行。称取样品0.2 g左右,首先在100℃下真空脱气4 h,液氮冷肼中进行N2吸附-脱附测定。催化剂比表面积采用BET法进行计算,孔体积和孔径根据BJH脱附法计算。
催化剂样品的红外光谱分析(FT-IR)和原位红外光谱分析(in-situIR)在美国NICOLET NEXUS 470型傅里叶红外光谱仪上进行。仪器分辨率为4 cm-1,波数范围400~4000 cm-1。FT-IR分析采用KBr压片法制样。CO2与乙酸锌的原位红外光谱测定操作过程如下:取一定量的无水乙酸锌充分研磨后制成自撑片,装入样品池中,用Ar吹扫并升温至160℃处理2 h;降温至100℃,将气氛切换为CO2进行吸附,保持30 min;再将气氛切换回Ar吹扫 1 h脱除物理吸附的CO2后,进行红外光谱分析并逐渐升高温度观察CO2化学吸附量的变化情况。PG与乙酸锌的原位红外光谱测定操作过程如下:首先,抽真空净化系统,升温至120℃并导入PG蒸气,测取无水PG蒸气的红外光谱;然后,称取一定量无水乙酸锌充分研磨后制成自支撑片,置于红外反应池中,真空条件下升温至120℃净化样品2 h后,将无水PG蒸气导入,用Ar吹扫1 h脱除物理吸附PG,并测定其红外光谱。在进行CO2或PG与乙酸锌的原位红外测定过程中,均以净化后的无水乙酸锌为背景。
1.4 CO2与PG合成PC反应操作
CO2与PG合成PC反应在内径为8 mm的固定床反应器中进行。具体操作过程如下:将2 ml Zn(OAc)2/AC催化剂装填入反应器恒温段,同时控制CO2在4.0 MPa条件下以一定流速通入,当催化剂床层升至预定反应温度后,用计量泵将溶剂乙腈和PG的混合液按一定流速通入反应器进行反应,液态反应产物及未反应的反应原料经冷却后进入储罐,每隔2 h取样进行定量分析。PC收率和选择性以PG为基准进行计算。
1.5 产物定量分析
反应液定量分析在北京北分瑞利分析仪器有限责任公司生产的SP3420A型气相色谱仪上进行。色谱柱为PEG20M,柱温采用程序升温控制,初温100℃,保持3 min,然后以10 ℃·min-1的速率升温到220℃,保持10 min;氢火焰检测器,温度220℃;气化室温度220℃。内标法定量,以正丁醇为内标物。
2 结果与讨论
2.1 反应体系的定性分析
以Zn(OAc)2/AC为催化剂,在PG与乙腈与CO2的摩尔比为 1:1.8:16、CO2压力 4.0 MPa、Zn(OAc)2/AC装填量2 ml、反应温度160℃和液空速0.9 h-1条件下,于固定床上进行了CO2与PG合成PC的反应。反应结束后,首先采用气相色谱对反应液进行了定量分析,得到PC的收率和选择性,分别为8.8%和27.7%。然后,采用GC-MS对反应液进行了定性分析,发现反应液中除了PG、PC、H2O和CH3CN外,还存在乙酰胺、1,2-丙二醇-2-乙酸酯、1,2-丙二醇二乙酸酯和一缩二丙二醇。据此推测主副反应如下。
主反应
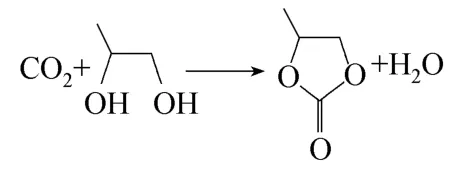
副反应

2.2 不同载体负载乙酸锌的催化性能
图1为不同载体对负载型乙酸锌催化性能的影响。不同载体负载乙酸锌催化剂上PC收率和选择性的次序均为:AC>SiO2>γ-Al2O3>MgO。酸性载体γ-Al2O3和碱性载体MgO的效果均不好。当以活性炭为载体时,PC的收率和选择性最高,分别为8.8%和27.7%。表1为不同载体制备负载型乙酸锌催化剂的比表面积及孔结构。比表面积的次序为Zn(OAc)2/AC>Zn(OAc)2/SiO2>Zn(OAc)2/γ-Al2O3>Zn(OAc)2/MgO,孔体积的次序为Zn(OAc)2/SiO2>Zn(OAc)2/γ-Al2O3>Zn(OAc)2/AC>Zn(OAc)2/MgO,平均孔径的次序为Zn(OAc)2/MgO>Zn(OAc)2/SiO2> Zn(OAc)2/γ-Al2O3>Zn(OAc)2/AC。可以看出,催化剂的比表面积对其催化性能的影响比较大,可能是由于比较大的比表面积有利于活性组分在催化剂表面的分散,以及反应原料与活性组分之间的充分 接触。
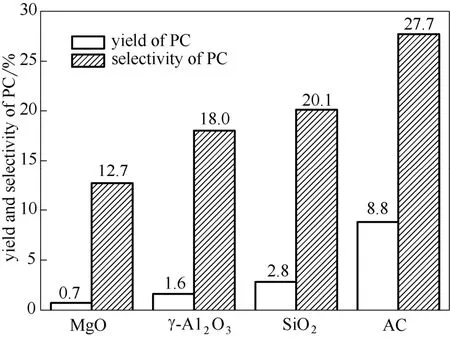
图1 载体对负载型乙酸锌催化性能的影响 Fig.1 Effect of supporter on catalytic performance of supported Zn(OAc)2catalysts reaction conditions: 160℃ , CO2pressure of 4.0 MPa, molar ratio of PG to acetonitrile to CO2=1:1.8:16, LHSV=0.9 h-1, Zn(OAc)2/AC volume of 2 ml

表1 不同载体制备负载型乙酸锌催化剂的比表面积和孔结构 Table 1 Specific surface area and pore structure of supported Zn(OAc)2catalysts prepared with different supporters
2.3 乙酸锌负载量的影响
在确定以活性炭为载体的基础上,制备了不同负载量的Zn(OAc)2/AC催化剂。首先对其进行了XRD分析,在XRD谱图中均未见到Zn(OAc)2的特征衍射峰,这可能是因为Zn(OAc)2单层分散在活性炭载体表面[13]。表2是不同负载量的Zn(OAc)2/AC催化剂的比表面积和孔结构测定结果。催化剂的比表面积和孔体积随着Zn(OAc)2负载量的增加逐渐减小,而平均孔径逐渐增大。表明活性组分Zn(OAc)2负载后会堵塞一部分活性炭的孔道,并且优先堵塞孔径较小的孔道,从而影响催化剂的比表面积。

表2 不同负载量Zn(OAc)2/AC催化剂的比表面积和孔结构 Table 2 Specific surface area and pore structure of Zn(OAc)2/AC catalysts with different loading
考察了Zn(OAc)2负载量对Zn(OAc)2/AC催化性能的影响,结果如图2所示。随着Zn(OAc)2负载量的提高,PC的收率和选择性均逐渐升高,当Zn(OAc)2的负载量增至40%(质量分数)时,PC的收率和选择性同时达到最大值,分别为8.8%和27.7%。继续增加Zn(OAc)2的负载量,PC的收率和选择性均略有降低。李国英等[12]研究发现,Zn(OAc)2在活性炭表面呈单层分散的负载量极限值为36%(质量分数),低于该极限值时催化剂的活性随负载量增加逐渐增大,高于该极限值后,Zn(OAc)2在催化剂表面呈多层覆盖,催化剂活性基本稳定。这与本研究结果基本一致。因此,确定Zn(OAc)2适宜的负载量为40%(质量分数)。

图2 Zn(OAc)2负载量对Zn(OAc)2/AC催化性能的影响 Fig.2 Effect of Zn(OAc)2loading on catalytic performance of Zn(OAc)2/AC reaction conditions: 160 , CO℃ 2pressure of 4.0 MPa, molar ratio of PG to acetonitrile to CO2=1:1.8:16, LHSV=0.9 h-1, Zn(OAc)2/AC volume of 2 ml
2.4 操作条件对PG与CO2合成PC反应的影响
2.4.1 液空速的影响 考察了液空速对CO2与PG反应合成PC反应的影响,结果见图3。在所考察范围内,液空速对PC收率和选择性的影响较为明显。随着液空速的增加,PC收率和选择性均呈现先升高后降低的变化趋势。当液空速为0.9 h-1时,PC的收率和选择性达到最高,分别为8.8%和27.7%。当液空速为1.2 h-1时,PG在催化剂床层的停留时间短,导致PC的收率和选择性较低。而当液空速为0.6 h-1时,PG在催化剂床层的停留时间较长,会导致一些副反应(如PC分解和聚合等)的加剧[9]。因此,选择0.9 h-1为适宜的液空速。

图3 液空速对PG与CO2合成PC反应的影响 Fig.3 Effect of LHSV on reaction of PG and CO2to PC reaction conditions: 160 , CO℃ 2pressure of 4.0 MPa, molar ratio of PG to acetonitrile to CO2=1:1.8:16, Zn(OAc)2/AC volume of 2 ml

图4 反应温度对PG与CO2合成PC反应的影响 Fig.4 Effect of reaction temperature on reaction of PG and CO2to PC reaction conditions: CO2pressure of 4.0 MPa, molar ratio of PG to acetonitrile to CO2=1:1.8:16, LHSV=0.9 h-1, Zn(OAc)2/AC volume of 2 ml
2.4.2 反应温度的影响 反应温度对PC合成反应的影响结果如图4所示。随着反应温度的升高,PC 的收率和选择性均呈现先增加后降低的变化趋势。采用Aspen估算了反应条件下PG与CO2合成PC的反应焓变,按照反应温度从低到高依次为-3.97、-3.87、-4.11、-4.68 kJ·mol-1。可见该反应为放热反应,虽然升高温度对反应平衡不利,但从动力学角度考虑,升高反应温度有助于反应速率的提高。在反应温度低于160℃时,PC收率随反应温度升高逐渐增大。推测是随反应温度升高,反应速率加快,从而PC收率逐渐增大。PC收率在反应温度为160℃时取得最大值,为8.8%,此时其选择性为27.7%。而PC选择性在170℃时取得最大值为33.4%,此时PC收率为6.5%。在PC的收率和选择性升高至最大值之后,继续升高反应温度,二者都有下降的趋势。一方面,反应温度升高,对反应平衡不利,导致PC收率降低;另一方面,根据热力学计算,反应体系中存在的PG与乙酸生成1,2-丙二醇二乙酸酯的副反应[7],属于吸热反应,温度升高有利于该副反应的发生;同时有文献[1-3]报道反应温度过高,会导致PG分子间脱水聚合和PC分解等副反应发生。上述原因共同导致了反应温度过高时PC收率和选择性的下降。170℃时PC的选择性相对于 160℃时提高了20.6%,而PC收率相对于160℃时降低了26.1%。综合考虑,确定PG与CO2合成PC的适宜反应温度为160℃。
2.4.3 原料配比的影响 考察了CO2和PG摩尔比对PC合成反应的影响,结果如图5所示。随着CO2和PG摩尔比的增加,CO2在反应体系中的浓度逐渐增加,有利于反应平衡向生成PC的方向移动,因此,PC的收率和选择性先是逐渐增大。CO2和PG的摩尔比较小时,可能是由于反应体系中CO2的浓度相对较低,导致原料PG参与副反应(如PG与乙酰胺或乙酸锌反应生成1,2-丙二醇-2-乙酸酯,PG与乙酸反应生成1,2-丙二醇二乙酸酯[7])的量较多,从而使得PC的收率和选择性较低。当CO2和PG的摩尔比为11时,PC的选择性达到最高为49.0%,此时PC的收率为6.3%。当CO2和PG摩尔比为16时,PC的收率达到最高,为8.8%,但PC的选择性只有27.7%。伴随着CO2和PG摩尔比从11增加至16,虽然PC的收率增加了2.5%,但其选择性却降低了21.3%。因此,综合考虑确定CO2和PG适宜的摩尔比为11。

图5 CO2与PG摩尔比对PG与CO2合成PC反应的影响 Fig.5 Effect of molar ratio of CO2to PG on reaction of PG and CO2to PC reaction conditions: 160 , CO℃ 2pressure of 4.0 MPa, molar ratio of PG to acetonitrile=1:1.8, LHSV=0.9 h-1, Zn(OAc)2/AC volume of 2 ml
2.5 乙酸锌催化机理的推测
2.5.1 PG与无水乙酸锌之间的作用分析 首先,采用原位红外技术对PG与无水乙酸锌的作用机理进行了分析,结果如图6所示。曲线a为PG蒸气的红外谱图。3329 cm-1处为PG中OH的伸缩振动吸收峰,2976 cm-1处为PG中CH3的反对称伸缩振动吸收峰,2936 cm-1处为PG中CH2的反对称伸缩振动吸收峰,CH3和CH2的对称伸缩振动以及CH的反对称和对称伸缩振动吸收峰重叠于2883 cm-1处[14]。曲线b为吸附PG的无水乙酸锌在脱除物理吸附PG后的红外图谱。在曲线b上可以看到CH3、CH2以及CH的反对称伸缩振动以及对称伸缩振动的吸收峰,而OH的伸缩振动吸收峰基本消失,说明PG可能通过OH与无水乙酸锌发生了某种化学作用。

图6 无水乙酸锌与PG原位反应红外谱图 Fig.6 FT-IR spectra of anhydrous zinc acetate interacted with PG
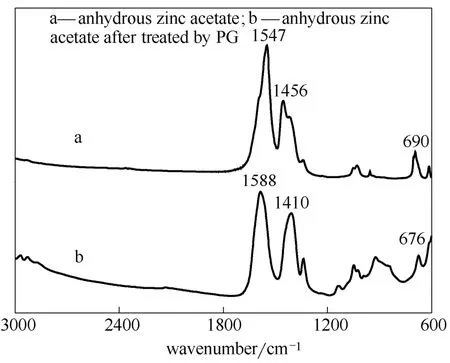
图7 经PG处理前后无水乙酸锌的红外谱图 Fig.7 FT-IR spectra of anhydrous zinc acetate before and after treated by PG
为了分析PG与无水乙酸锌作用后无水乙酸锌自身的变化情况,设计了如下实验:将PG与无水乙酸锌加入高压反应釜,用N2置换釜内空气后,升温至160℃并搅拌2 h。实验结束后,将过滤所得的乙酸锌经洗涤、真空干燥处理后进行红外分析,结果如图7所示。对于无水乙酸锌,1547 cm-1和1456 cm-1处分别对应于乙酸根反对称伸缩振动和对称伸缩振动的吸收峰。两吸收峰之间的间距为91 (<100 cm-1),据此可知无水乙酸锌为典型的双齿配位结构[14]。经PG化学处理后,乙酸根的反对称伸缩振动吸收峰向高频移动,而对称伸缩振动吸收峰向低频移动,二者间的间距增大至178 cm-1(>160 cm-1),推测经PG处理后的乙酸锌变为单齿配位结构[13]。690 cm-1和676 cm-1处的吸收峰分别对应于经PG化学处理前后乙酸锌中ZnO键的吸收峰[15]。推测可能是经PG处理后,Zn2+上连接的含氧基团发生变化,从而使得Zn—O键红外吸收峰的位置发生了偏移。
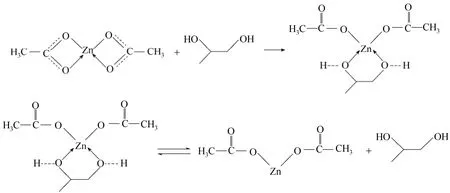
Baba等[16]研究发现甲醇通过其OH上的氧与Zn2+配位可以使乙酸锌由双齿配位结构转化为单齿配位结构。再结合以上红外分析结果,推测PG与无水乙酸锌的作用机理为:PG中两个OH上的氧分别进攻无水乙酸锌中的Zn2+,使Zn2+与乙酸根中氧之间的配位键打开,与PG中两个OH上的氧配位,形成一种新的配位化合物。该配位化合物可能不稳定,导致与Zn2+配位的PG从无水乙酸锌表面解离。因此,检测到的无水乙酸锌为单齿配位结构。具体反应式如下
2.5.2 CO2与无水乙酸锌之间的作用分析 采用原位红外分析技术对CO2在无水乙酸锌表面的化学吸附情况进行了分析,结果如图8所示。图中所标注的时间为CO2物理吸附结束后Ar吹扫的总时间。可以看出,随着温度的升高以及时间的延长,无水乙酸锌表面吸附的CO2逐渐减少。当温度升至160℃、吹扫时间延长至110 min时,催化剂表面仍吸附有一定量的CO2,表明CO2与无水乙酸锌之间有一定的作用力存在。但吸附前后CO2红外吸收峰的位置没有发生改变,可能是由于CO2在无水乙酸锌上的化学吸附比较弱。
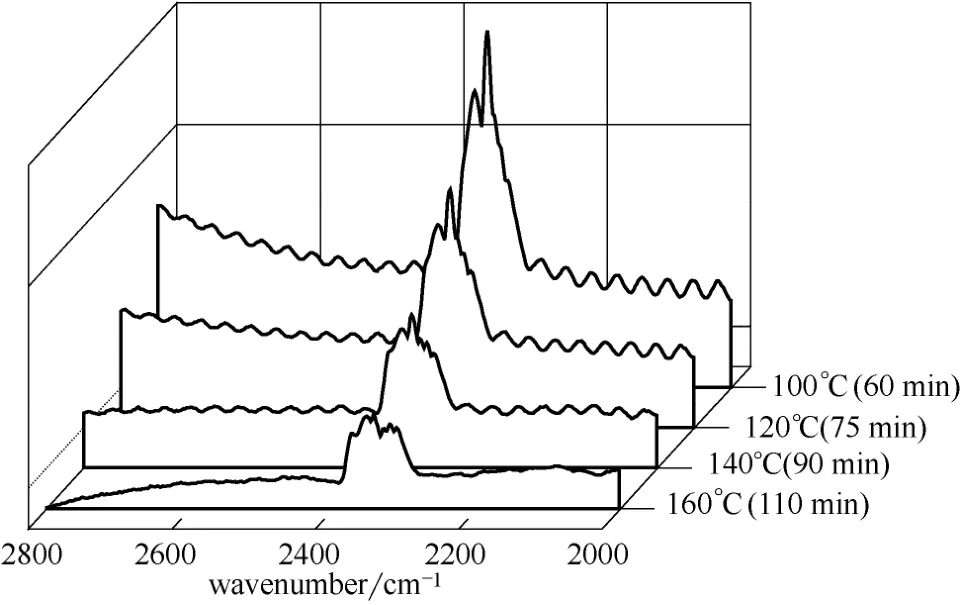
图8 无水乙酸锌与CO2原位反应红外谱图 Fig.8 FT-IR spectra of anhydrous zinc acetate interacted with CO2
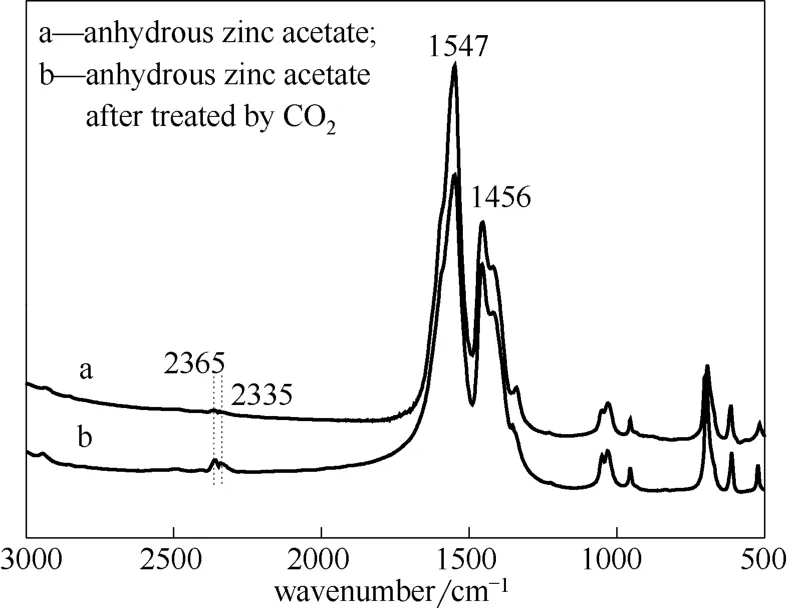
图9 经CO2处理前后无水乙酸锌的红外谱图 Fig.9 FT-IR spectra of anhydrous zinc acetate before and after treated by CO2
为了分析CO2与无水乙酸锌作用后催化剂的变化情况,分别对CO2处理前后的乙酸锌进行了红外光谱分析,结果如图9所示。CO2对无水乙酸锌的处理方法为:将乙腈和无水乙酸锌加入高压反应釜,在CO2初压3.0 MPa、160℃条件下处理2 h,处理后的乙酸锌经过滤、洗涤和干燥后进行红外分析。图中1547 cm-1和1456 cm-1处分别对应于乙酸根反对称伸缩振动和对称伸缩振动的吸收峰,2365 cm-1和2335 cm-1处分别归属为CO2反对称伸缩振动的P支和Q支[14]。对比CO2处理前后乙酸锌的红外谱图可知,经CO2处理后,有CO2吸附在催化剂表面,可能是由于吸附比较弱,乙酸根的反对称伸缩振动和对称伸缩振动吸收峰没有明显的位移或宽化现象出现,只是相对强弱发生一定变化。
2.5.3 无水乙酸锌催化机理的推测 PG和CO2分别与无水乙酸锌作用的研究结果表明,CO2与无水乙酸锌之间的作用力比较弱,主要是无水乙酸锌对PG起到活化作用。据此推测无水乙酸锌催化PG与CO2合成PC的反应机理如图10所示。第1步,PG中两个OH上的氧分别进攻具有双齿结构乙酸锌(species 1)中的Zn2+,使Zn2+与乙酸根中氧之间的配位键打开,与PG中两个OH上的氧配位,形成一种新的配位化合物species 2;第2步,配位化合物species 3与species 2为互变异构体,species 3很容易失去两分子乙酸,从而转化为species 4;第3步,化合物species 4中两个带负电的醇氧基负离子分别进攻CO2的羰基碳原子,使碳氧双键打开,氧原子带负电荷,两个带负电的氧原子与Zn2+以离子键形式结合形成化合物species 6;第4步,化合物species 6与第2步中生成的乙酸作用生成化合物species 7,同时乙酸根与Zn2+结合为乙酸锌;第5步,化合物species 7中连在同一个碳上的两个OH不稳定,脱去一分子水生成目标产物PC。
综上,与Zhao等[7]在高压反应釜上进行的无水乙酸锌均相催化PG与CO2合成PC反应结果(PC收率为12.3%)相比,在固定床上得到的PC收率仍然较低。采用Aspen估算了反应条件下PG与CO2合成PC的反应平衡常数,在所考察的反应温度范围内该反应的反应平衡常数均较小,按照反应温度从低到高反应平衡常数依次为1.07×10-3、1.01×10-3、9.52×10-4和9.04×10-4。因此,反应平衡常数较小是PC收率较低的原因之一。此外,还可能有两方面原因:一方面,受CO2饱和蒸气压的限制,普通固定床上的反应压力较低;另一方面,结合推测的反应机理分析可知,原料PG与活性组分乙酸锌接触并被活化是反应进行的关键,负载型乙酸锌可能不如均相乙酸锌与PG接触充分。根据上述分析,有3种方法有可能能够改善在固定床上进行PC合成反应的反应效果:①在反应体系中加入脱水剂,打破反应平衡,促进平衡向右移动;②提高反应压力,如采用超临界固定床反应器;③改进催化剂的制备方法,提高乙酸锌在催化剂表面的分散度,使催化剂有更多的活性中心可以活化PG。

图10 乙酸锌催化PG与CO2合成PC反应机理 Fig.10 Possible mechanism for PC synthesis from CO2and PG catalyzed by zinc acetate
3 结 论
(1)以负载型乙酸锌为催化剂,首次在固定床反应器上实现了CO2与PG合成PC的反应。以活性炭为载体,负载量为40%(质量分数) 的负载型乙酸锌催化性能最好。结合表征结果发现,大比表面积的中性载体因有利于乙酸锌在其表面的分散而具有较好的效果。
(2)Zn(OAc)2/AC催化固定床上CO2与PG合成PC反应的适宜反应条件为:PG、乙腈和CO2的摩尔比为 1:1.8:11,CO2压力 4.0 MPa,Zn(OAc)2/AC装填量2 ml,反应温度160℃和LHSV=0.9 h-1。在此条件下,PC的收率和选择性分别为6.3%和49.0%。研究结果表明,在连续反应装置上进行CO2与PG合成PC反应是可行的。
(3)采用原位红外技术结合设计实验研究了无水乙酸锌分别与CO2和PG之间的相互作用,发现CO2与乙酸锌之间有化学吸附存在,但作用比较弱,而乙酸锌与PG之间的化学吸附比较强,能够活化PG。据此推测了乙酸锌催化CO2与PG合成PC的反应机理。
[1] Tomishige K, Yasuda H, Yoshida Y, Nurunnabi M, Li B, Kunimori K.Novel route to propylene carbonate: selective synthesis from propylene glycol and carbon dioxide [J].Catalysis Letters, 2004, 95 (1/2): 45-49
[2] Tomishige K, Yasuda H, Yoshida Y, Nurunnabi M, Li B, Kunimori K.Catalytic performance and properties of ceria based catalysts for cyclic carbonate synthesis from glycol and carbon dioxide [J].Green Chemistry, 2004, 6 (4): 206-214
[3] Du Y, Kong D L, Wang H Y, Cai F, Tian J S, Wang J Q, He L N.Sn-catalyzed synthesis of propylene carbonate from propylene glycol and CO2under supercritical conditions [J].Journal of Molecular Catalysis A:Chemical, 2005, 241 (1): 233-237
[4] Du Y, He L N, Kong D L.Magnesium-catalyzed synthesis of organic carbonate from 1,2-diol/alcohol and carbon dioxide [J].Catalysis Communications, 2008, 9: 1754-1758
[5] Chen Hong (陈鸿), Zhao Xinqiang (赵新强), Wang Yanji (王延吉).Synthesis of propylene carbonate from carbon dioxide and 1,2-propylene glycol over potassium carbonate catalyst [J].Petrochemical Technology(石油化工), 2005, 34 (11): 1037-1040
[6] Chen Hong (陈鸿), Zhao Xinqiang (赵新强), Wang Yanji (王延吉).Synthesis of propylene carbonate from carbon dioxide and 1,2-propylene glycol over supported potassium carbonate catalyst [J].Journal of Chemical Engineering of Chinese Universities(高校化学工程学报), 2006, 20 (5): 734-739
[7] Zhao Xinqiang, Sun Na, Wang Shufang, Li Fang, Wang Yanji.Synthesis of propylene carbonate from carbon dioxide and 1,2-propylene glycol over zinc acetate catalyst [J].Industrial & Engineering Chemistry Research, 2008, 47 (5): 1365-1369
[8] Huang Shiyong (黄世勇), Zhao Ning (赵宁), Guo Qiwen (郭启文), Li Junping (李军平), Wei Wei (魏伟), Sun Yuhan (孙予罕).Synthesis of propylene carbonate from carbon dioxide and 1,2-propylene glycol with organic base catalyst [J].Petrochemical Technology(石油化工), 2007, 36 (6): 601-604
[9] Huang S Y, Ma J, Li J P, Zhao N, Wei W, Sun Y H.Efficient propylene carbonate synthesis from propylene glycol and carbon dioxide viaorganic bases [J].Catalysis Communications, 2008, 9 (2): 276-280
[10] Huang Shiyong (黄世勇), Ma Jun (马珺), Zhao Ning (赵宁), Dong Manxiang (董满祥), Wei Wei (魏伟), Sun Yuhan (孙予罕).Synthesis of propylene carbonate from carbon dioxide and 1,2-propylene glycol with ferric chloride catalyst [J] Petrochemical Technology(石油化工), 2007, 36 (3): 248-251
[11] Huang S Y, Liu S G, Li J P, Zhao N, Wei W, Sun Y H.Modified zinc oxide for the direct synthesis of propylene carbonate from propylene glycol and carbon dioxide [J].Catalysis Letters, 2007, 118 (3/4): 290-294
[12] Li Guoying (李国英), Chen Shu (陈曙), Wang Yuqing (王玉庆), Yu Boling (于伯龄).Relation between the activity and structure of zinc acetate/activated carbon catalyst [J].Petrochemical Technology(石油化工), 1984, 13 (10): 649-654
[13] Mei Yan (梅燕), Nie Zuoren (聂祚仁), Wang Wei (王为).Application of IR spectrum to the research on coordination mechanisms of organic carboxylic acid during homogeneous precipitation [J].Spectroscopy and Spectral Analysis(光谱学与光谱分析), 2007, 27 (2): 254-258
[14] Weng Shifu (翁诗甫).Fourier Transform Infrared Spectrometer (傅里叶变换红外光谱仪)[M].Beijing: Chemical Industry Press, 2005
[15] Ma Dan (马丹), Wang Guirong (王桂荣), Wang Yanji (王延吉), Zhao Xinqiang (赵新强).Application of IR spectrum to the research on mechanism of synthesis of 2,4-toluene dicarbamate catalyzed by zinc acetate [J].Spectroscopy and Spectral Analysis(光谱学与光谱分析), 2009, 29 (2): 331-335
[16] Baba T, Kobayashi A, Kawanami Y, Inazu K, Ishikawa A, Echizenn T, Murai K, Aso S, Inomata M.Characteristics of methoxycarbonylation of aromatic diamine with dimethyl carbonate to dicarbamate using a zinc acetate catalyst [J].Green Chemistry, 2005, 7 (3): 159-165
