盐酸氟桂利嗪的合成工艺改进
2015-08-19陈连锋张丁王风云夏明珠雷武朱其军
陈连锋,张丁,王风云,夏明珠,雷武,朱其军
(1南京理工大学工业化学研究所,江苏 南京 210094;2盐城格瑞茵化工有限公司,江苏 盐城 224400)
盐酸氟桂利嗪系钙通道阻断剂,是一种双氧化哌嗪衍生物,能防止因病理性钙超载而造成的细胞受损[1],近十年,该产品相关文献的报道并不多,但因为其在偏头痛、耳鸣、椎基底动脉[2]等疾病的治疗上有良好的功效和安全性,所以很有必要作进一步的研究。该产品由Janssen于1970年[3]首次合成,经过一系列的临床试验,在欧洲的一些国家于20世纪80年代初上市。
目前已有多篇文献报道盐酸氟桂利嗪的合成,其合成方法主要可分两大类[4-5]。①以对氟苯甲酸和二氯亚砜为原料,此工艺优点在于反应条件温和,缺点是原料对氟苯甲酸有强腐蚀性,需定制,且反应周期长,收率低,仅为18%左右。②以氟苯和三氯化铝为原料,此工艺和①相比,缺点是使用一氧化碳和溴素作原料,且需在高压釜中反应,反应条件苛刻,副反应多,收率低,总收率仅13%左右。
针对以上问题,作者在参考文献[6-9]的基础上,对整个工艺路线进行了改进。以氟苯和肉桂醇为起始原料,氟苯经傅克烷基化、还原、溴代得到双(4-氟苯基)溴甲烷;肉桂醇经氯代,再和哌嗪反应得到肉桂基哌嗪,再和双(4-氟苯基)溴甲烷反应得到最终产品。此工艺与原有工艺相比,减少了一步反应,避免了原料一氧化碳[10-11]的使用,在合成双(4-氟苯基)溴甲烷时,使用NBS代替溴素,避免了使用溴素时的易挥发和强腐蚀性。最后一步合成采用三乙胺/二氯乙烷反应体系,四丁基溴化铵作相转移催化剂,和以往的碳酸钾/丙酮体系相比,无需反应7h[4],3h已反应完全,且结晶时不需要加入晶种,反应条件温和且操作简便,提高了最终产率,具体的合成路线如图1。
1 实验部分
1.1 主要试剂与仪器
肉桂醇、氢氧化钾、硼氢化钠、乙醇(95%)、四氢呋喃、二氯亚砜、碳酸氢钠、甲苯、哌嗪、三氯化铝、四氯化碳、N-溴代丁二酰亚胺、偶氮二异丁腈、二氯乙烷、盐酸、无水硫酸镁,所用试剂均为CP,南京化学试剂有限公司。
X-5显微熔点测定仪,巩义市予华仪器有限责任公司;P230高效液相色谱仪,大连依利特分析仪器有限公司;TENSOR-27型傅里叶快速红外光谱仪,瑞士Bruker公司;MAT-8200质谱仪,Finnegan MAT公司;Bruker AVANCEⅢ400MHz核磁共振波谱仪,瑞士Bruker公司。
1.2 合成步骤
1.2.1 4,4'-二氟二苯甲酮的制备[12]
向250mL四口烧瓶中加入CCl4(80g , 0.52mol),AlCl3(34.6g,0.26mol),PEG-400(4g,0.01mol),冰浴下搅拌降温至10℃,向其中缓慢滴加氟苯(24g,0.25mol),15min滴毕,45℃搅拌2h。抽滤,向滤液中缓慢加入100mL冰水,蒸出未反应的氟苯和四氯化碳,趁热将烧瓶中的剩余物倒入200 mL冰水中,立刻析出大量的深红色晶体,抽滤,将晶体溶于适量的二氯乙烷,再加入0.5g活性炭脱色,保温1h,趁热抽滤,冷却结晶,过滤,滤饼用少量无水乙醇淋洗,干燥,得片状白色晶体24.2g,收率88.6%,熔点103~104℃(文献[3]:102~ 105℃)。
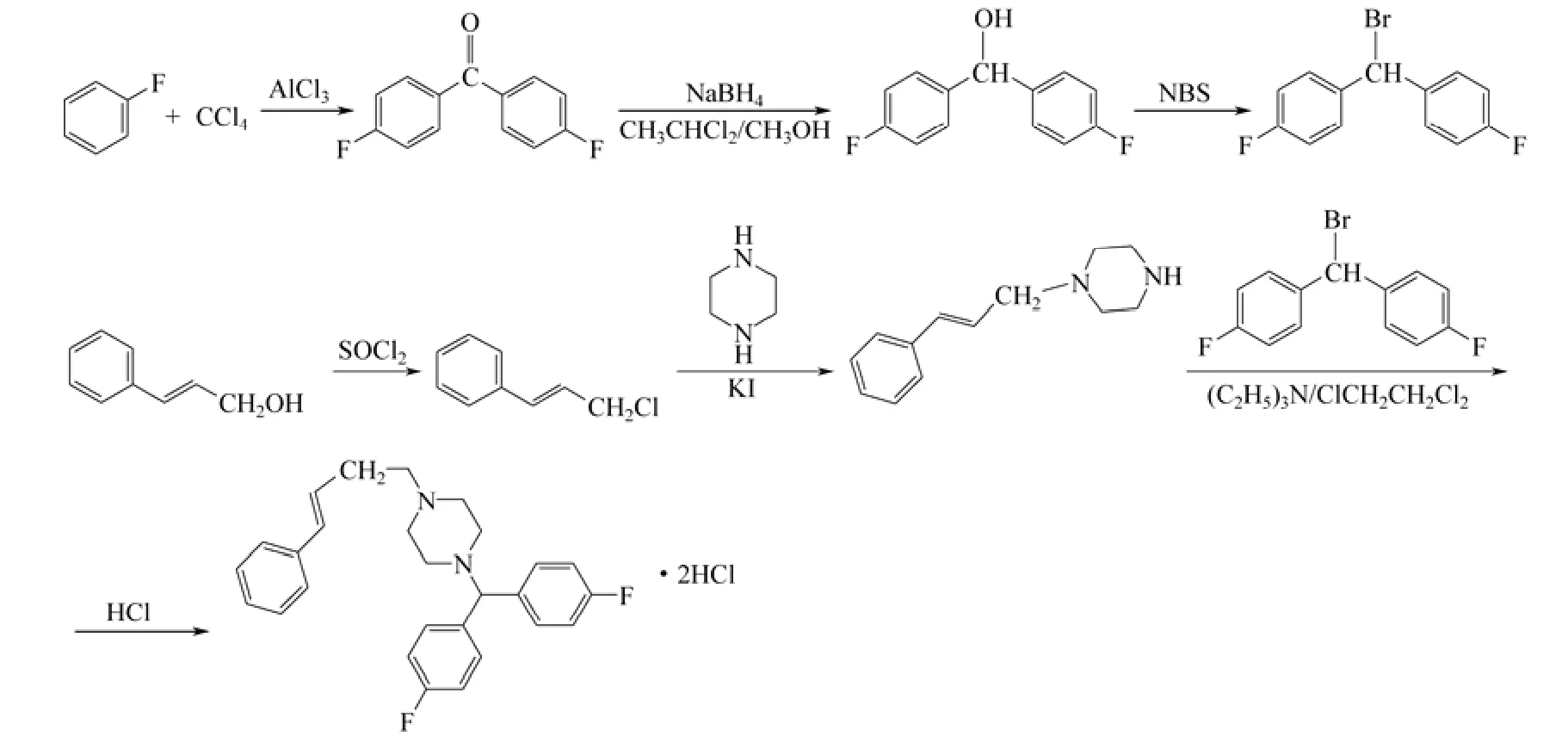
图1 盐酸氟桂利嗪的合成路线
1.2.2 4,4'-二氟二苯甲醇的制备[13]
向250mL四口烧瓶中加入甲醇(20g,0.625mol),硼氢化钠(2.5g,0.066mol),冰浴下搅拌降温至5℃,向烧瓶中缓慢滴加4,4'-二氟二苯甲酮(28.8g,0.132mol)的二氯乙烷(30.9g,0.31mol)溶液,滴毕,升至50℃保温反应,TLC(展开剂石油醚∶乙酸乙酯=15∶1,体积比,下同)跟踪反应至反应结束,加入稀盐酸淬灭,静置分层,水层用20mL×2二氯乙烷萃取,合并有机层,加入冰水250mL,搅拌15min,抽滤,干燥,得白色固体26.8g,收率92.1%,熔点43~44℃(文献[7]:43~45℃)。
1.2.3 双(4-氟苯基)溴甲烷的制备[5,14]
将4,4'-二氟二苯甲醇(18g,0.08mol),NBS(14.2g,0.08mol),AIBN(0.40g,0.0024mol)和二氯乙烷(36g,0.35mol)加入到250mL四口烧瓶中, 搅拌下升温至80℃,TLC(石油醚∶乙酸乙酯=15∶1)跟踪反应至反应结束,冷却,过滤,母液分别用饱和碳酸氢钠溶液和饱和食盐水洗涤数次,静置分层,有机层用无水硫酸镁干燥,旋干溶剂二氯乙烷,得黄色固体20.1g,收率88.8%。
1.2.4 肉桂基氯的制备[15]
向250mL四口烧瓶中加入二氯亚砜(55.9g,0.47mol),二氯乙烷(35g,0.35mol),搅拌冰浴下降温至5℃左右,向烧瓶中缓慢滴加苯基丙烯醇(32.5g,0.24mol)和二氯乙烷(35g,0.35mol)的混合溶液,30min滴毕,升至70℃保温反应,TLC(展开剂石油醚∶乙酸乙酯=3∶1)跟踪反应至反应结束。降至室温,加入冰水150mL除去未反应的二氯亚砜,搅拌20min,静置,分层,再用50mL(8.6%)饱和碳酸氢钠溶液洗涤,静置,分层,用30mL×2二氯乙烷萃取水层,合并有机层,加无水硫酸镁干燥,抽滤,蒸馏除去溶剂二氯乙烷,得淡黄色液体34.6g,收率95.6 %。
1.2.5 肉桂基哌嗪的制备[7,15]
向250mL四口烧瓶中加入无水哌嗪(36.7g,0.43mol),KI (38.4g,0.23mol),甲苯(60g,0.65mol)和乙醇(30g,0.65mol),搅拌冰浴下降温至10℃,向烧瓶中缓慢滴加肉桂基氯(15.5g,0.10mol)和乙醇(30g,0.65mol),30 min滴毕,升温至50℃保温,TLC(展开剂石油醚∶乙酸乙酯=10∶1)跟踪反应至结束。搅拌降温,抽滤,向滤液中加入100mL冰水去除未反应的哌嗪,用40mL×2二氯乙烷萃取,合并有机层,用质量分数10%的稀盐酸调节pH值至5~6,静置分层,向水层中加入质量分数10%的NaOH调节pH值至9~10,用40mL×2二氯乙烷提取,合并有机层,加入适量无水硫酸镁干燥,旋干二氯乙烷,得深红色黏稠液体11.4g,收率56.2%。
1.2.6 盐酸氟桂利嗪的制备[16-19]
向250mL四口烧瓶中加入双(4-氟苯基)溴甲烷(25g,0.089mol),二氯乙烷(50g,0. 51 mol),室温搅拌使其溶解,加入三乙胺(18.0g,0.178mol),四丁基溴化铵(0.3g,9.3×10-4mol),冰浴下降温至10℃,向其中滴加肉桂基哌嗪(18.4g,0.091mol),15min滴毕,室温搅拌3h,加入100mL水稀释反应液,静置分层,水层用20mL×2二氯乙烷萃取,合并有机层,滴加稀盐酸调节pH值至5~6,振荡,静置分层,向水层中加入稀NaOH溶液调节pH值至9~10,加入30mL×2二氯乙烷萃取,分液,合并有机层,加入0.5g×2活性炭脱色30min,抽滤,加入稀盐酸调节pH值至5~6,旋干溶剂,得淡黄色固体,再用正丁醇重结晶,抽滤,真空干燥得白色固体粉末36.5g,收率76.0%,纯度99%以上,熔点188~189℃(文献[8]188~190℃)。产物结构经红外、质谱表征确定,见图2、图3。由图2可见,IR:3420cm-1(—C=C—),2999cm-1(苯环),2953cm-1(—CH2—),1511cm-1(苯环面内弯曲振动),1440cm-1(—C—N—)744cm-1(苯环面外弯曲振动)。由图3可见质谱表征结果:MS(m/z:405.1+H+)。
2 结果与讨论
2.1 4,4'-二氟二苯甲酮的制备
2.1.1 反应时间对收率的影响
其他反应条件同1.2.1节,仅改变反应时间,考查反应时间对收率的影响,结果见表1。

图2 盐酸氟桂利嗪的红外谱图
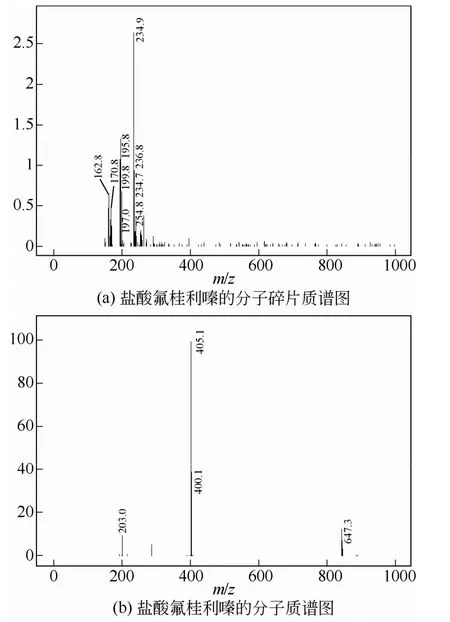
图3 盐酸氟桂利嗪的质谱图
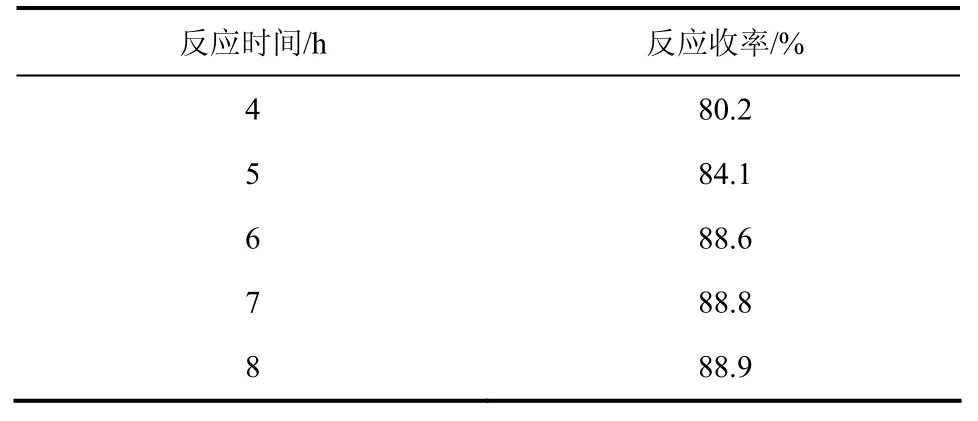
表1 反应时间对收率的影响
由表1可知,当反应时间由7h增加到8h时,产率增幅趋于平稳,从能耗角度考虑,6h为最适宜反应时间。
2.1.2 4,4'-二氟二苯甲酮的精制
氟是吸电子基,不利于傅克反应的进行,所以只有适当升高温度和延长反应时间,但这样就不可避免地引入副产物,此副产物也是影响最终产品纯度的主要杂质,因此采用脱色和重结晶方法对其提纯,用甲醇、乙醇、二氯乙烷和甲苯分别对其重结晶;结果表明,二氯乙烷的效果最好,所得产品纯度和收率最高。
2.2 4,4'-二氟二苯甲醇的制备
分别使用Zn粉和硼氢化钠进行还原,发现用Zn粉后处理步骤繁琐,且活化的Zn粉比较危险,抽干时在空气中就会自燃,用硼氢化钠效果更好,反应条件温和,后处理简便,产率也较高。进一步 做了硼氢化钠的单因素实验,结果见表2。

表2 硼氢化钠对反应收率的影响
由表2可知,当硼氢化钠的摩尔比值由0.7增加到0.8时,产率增幅趋于平稳,因此硼氢化钠的最佳用量为n(硼氢化钠)/n(4,4'-二氟二苯甲酮)=0.6。
2.3 双(4-氟苯基)溴甲烷的制备
4,4'-二氟二苯甲醇的溴代是按SN1机理进行的亲核取代反应,分别使用溴素、NBS、氢溴酸、氢溴酸的硫酸溶液进行溴代,结果见表3。
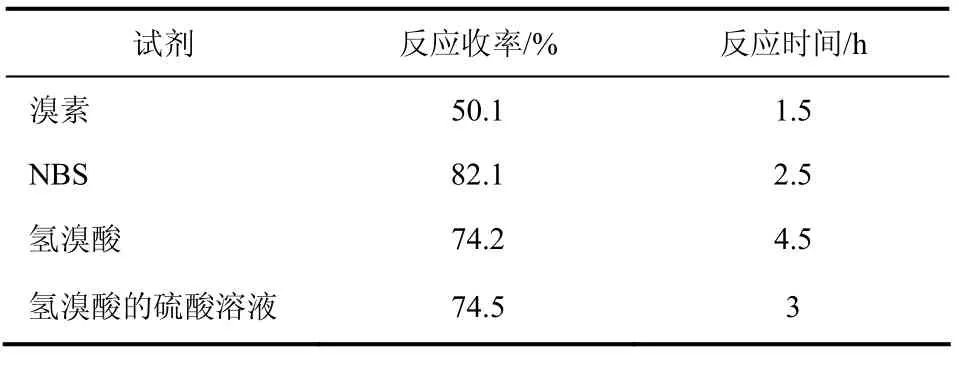
表3 溴代试剂的选择对反应收率的影响
由表3可知,虽然用溴素反应时间最短,但收率低,仅50%;选用氢溴酸的硫酸溶液溴代时反应时间比氢溴酸短,因为仲醇的亲核取代反应是按SN1机理进行的,可能原因是硫酸的加入更促进了碳正离子中间体的生成,但反应收率和使用氢溴酸时相当;选用NBS时收率最高,反应时间适中,综合反应时间和反应收率,选用NBS作溴代试剂最 适宜。
2.4 肉桂基氯的制备
分别使用二氯亚砜和浓盐酸进行氯代,发现用浓盐酸反应温度高,反应时间长,用二氯亚砜效果更好,产物收率更高,进一步做了二氯亚砜的单因素实验,结果如表4所示。由表4可知,随着二氯亚砜用量的增加,产物的收率也在增加,当二氯亚砜用量摩尔比值由2.2增加到2.4时,产物收率趋于平稳,因此二氯亚砜的最佳用量为n(二氯亚砜)/n(肉桂醇)=2.0。
2.5 肉桂基哌嗪的制备
由于哌嗪含有两个仲氨基,另一个仲氨基也会和肉桂基氯发生反应,因此,加大哌嗪的用量可以 减少甚至避免副反应的发生,哌嗪的用量为肉桂基氯的3~4倍为宜。

表4 二氯亚砜的用量对反应的影响
2.6 盐酸氟桂利嗪的合成
盐酸氟桂利嗪的合成分别采用了两种方法进行。第一种方法是以丙酮为溶剂,碳酸钾作缚酸剂,该方法反应周期长,需反应7h,结晶时需要加入晶种助结晶,操作繁琐,且产率低,只有50%左右;第二种方法是以二氯乙烷为溶剂,三乙胺作缚酸剂,四丁基溴化铵做相转移催化剂,反应条件温和,反应周期短,产率高达88.6%。因此,选用第二种方法,并且对此做了5组重复性实验,结果见表5。
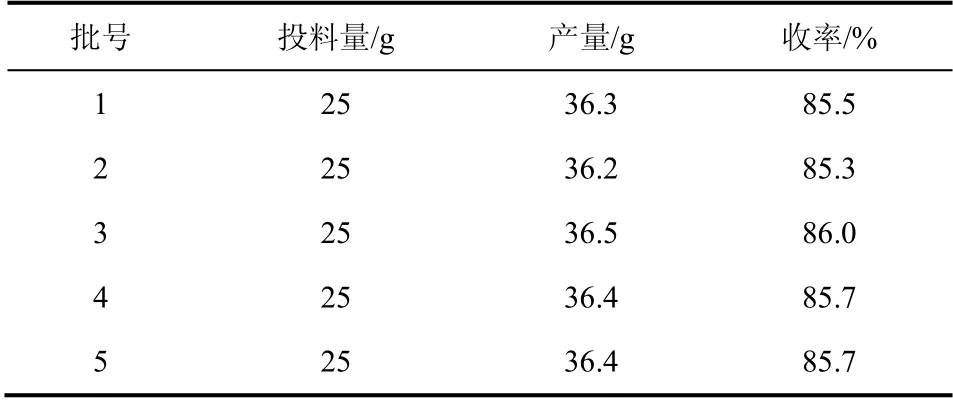
表5 盐酸氟桂利嗪的收率
3 结 论
以氟苯和四氯化碳为起始原料,三氯化铝为催化剂,经傅克烷基化、还原、溴代3步反应制得双(4-氟苯基)溴甲烷,肉桂醇经氯代、取代制得肉桂基哌嗪,再与双(4-氟苯基)溴甲烷反应制得盐酸氟桂利嗪,此工艺与文献[4]报道相比减少了一步,避免了一氧化碳的使用,且使用NBS代替溴素,操作简便,最后一步反应采用三乙胺/二氯乙烷的反应体系,四丁基溴化铵作相转移催化剂,反应时间由10h[4]缩短到3h,降低了能耗,减少了成本,总收率达30.0%。此工艺原料成本低廉,各步反应条件温和,步骤简便,具有很高的应用前景。
[1] Harada K,Nakamura A,Dobashi R. Effects of lomerizine,a new Ca2+entry blocker,on the contractile response of isolated canine cerebral and peripheral arteries[J].Journal of Veterinary Pharmacology and Therapeutics,1997,12(3):167-177.
[2] 欧阳青,李劲图,钟俊凯,等. 脑心通联合盐酸氟桂利嗪(西比灵)治疗椎基底动脉供血不足的疗效[J]. 中国老年学杂志,2012,32(13):66-67.
[3] 王泽民. 当代结构药物全集[M]. 北京:科学技术出版社,1991:4520-4523.
[4] 迟传金,孙庆,刘懋勤. 氟桂嗪的合成[J]. 上海医科大学学报,1987,14(4):314-315.
[5] 王立升,李敬芬,周天明. 氟桂利嗪的合成工艺研究[J]. 中国医药工业杂志,1987,28(10):438-440.
[6] 常秀娟,白仁才. 对氟桂利嗪有关物质检查方法的改进[J].山东医药工业,1998,4(3):78-79.
[7] 陈夏英. 脑益嗪的合成[J]. 医药工业,1982 ,4(10):4-5.
[8] 姚凤鸣,刘素梅. 脑益嗪合成方法的改进[J]. 医药工业,1984,5(4):41-42.
[9] 周晓东. 4,4,-二氟二苯甲酮的合成研究[J]. 精细石油化工进展,2004,5(5):72-73.
[10] Kyoko O,Toshisumi M,Akito W. Method for producing cyclohexanedione:JP,2001342163 A[P]. 2001-11-07.
[11] 李冰,张天永,池立峰,等. 2-取代间苯二酚化合物的合成[J]. 天津大学学报,2008,41(8):23-24.
[12] Ayyangar N R.(Trichloromethyl) benzene:A versatile reagent for the prepa ration of substituted benzophenones[J].Synthesis-Stuttgart,1991,5(4):322-324.
[13] Scott S C,Michael H K. A regioseleetive synthesis of 2,4-dialkyl resorcin ols[J].Tetrahedron Letters,2005,6(10):1731-1734.
[14] 江毓瑞,张明强,高咏新,等. 4,4’-二氟苯基氯甲烷合成[J]. 中国医药工业杂志,1985,16(8):34-36.
[15] 王国喜,孙保平,李淑君. 用哌嗪二盐酸盐合成肉桂基哌嗪[J]. 精细石油化工,2001,1(1):56-57.
[16] 项曼文,莫芬珠,张世春,等. 氟苯桂嗪的合成[J]. 南京药学院学报, 1984, 15(2):50-52.
[17] 李树有. 相转移技术及其在药物合成中的应用[J]. 山西化工,2001,3(1):45-46.
[18] 龚子东,孟祥涛,张香菊,等. 盐酸氟桂利嗪的改进措施[J]. 河南大学学报,2005, 24(3):3-4.
[19] 鲍春和,陈子明,黄超伦,等. 氟桂利嗪衍生物的合成[J]. 中国医药工业杂志,1991,22(5):211-212.
