构筑手性金属有机骨架的方法及其在不对称催化中的应用
2015-08-19潍坊学院化学化工与环境工程学院山东潍坊261061
(潍坊学院化学化工与环境工程学院,山东 潍坊 261061)
手性化合物作为医药、农药、香料以及功能材料等的前体、中间体或最终产物,在精细化工生产中占有极为重要的地位。获得单一对映体化合物的方法及其应用的研究成为了当代化学研究的前沿和热点之一[1-2]。设计合成具有手性对映体选择性、手性催化以及手性分离的手性沸石和相关的多孔材料具有非常重要的意义,然而关于这方面的报道还 较少[3]。
鉴于大孔沸石和分子筛在药物、助剂生产和分离过程中不可替代的重要作用,手性沸石和手性分子筛的合成一直是广大科研工作者不懈努力的目标。目前在合成大孔沸石和分子筛方面已经有了很大的突破,但是至今仍没有合成出手性沸石和手性分子筛。近年来,一种由有机配体与金属离子通过自组装过程形成的具有周期性网络结构的金属有机骨架晶体材料(metal-organic framework,MOF)引起了催化学者的极大重视[4-5]。它是由金属离子或金属节点与多齿有机配体(如对苯二甲酸、均苯三甲酸)自组装而成的配位聚合物,且能够在空间上形成一维、二维或三维的多孔结构的晶体材料[6-7]。制备MOF的金属离子和有机配体丰富多样,可以根据材料的性能,如官能团、孔道的尺寸和形状等加以选择。金属主要是以过渡金属(如Zn、Cu、Co)以及镧系金属元素等为主。常用的有机配体为含有N、O 等能提供孤对电子的刚性配体,如多羧酸、多磷酸、多磺酸、联吡啶、嘧啶等。与分子筛及沸石相比,同样具有微孔特征的MOF材料不仅可以在温和的条件下合成,而且通过修饰有机配体可以改变其物理和化学性质,通过选择恰当的建筑模块可以得到手性MOF。
早期有关MOF的综述主要集中在材料的制备、表征以及吸附和储氢等应用上[8-10],最近有几篇综述分别按在MOF上创立催化活性位的方法[11]、MOF催化的反应类型[1]、不对称催化反应[12-13]以及不对称催化活性位[14-15]进行了评述。本文重点概括和评述了创立手性MOF的方法,并讨论各自的优缺点,详细介绍手性MOF在不对称催化反应中的应用,指出MOF在不对称催化领域需要重视的问题和未来的主要研究方向,以期对MOF在不对称催化领域的研究和开发提供参考。
1 构筑手性MOF的方法
在金属有机骨架孔道中锚装或嫁接具有手性催化性能的配体、有机分子或有机金属化合物,制备的具有手性孔道结构或其孔道结构由某些具有手性特征的结构单元构成的MOF材料,均具有手性(不对称)催化与对映体选择分离材料的开发与应用前景,这对目前的催化领域来讲是很重要的一个前沿方向,因此有关手性MOF的合成与组装问题就显得非常重要。合成手性MOF的方法概括起来主要有4种方法,即①非手性物质在晶体生长过程中自组装形成具有螺旋结构的手性MOF;②使用手性化合物来诱导合成具有手性特征的手性MOF;③通过手性有机基团与金属离子配位将手性成分嵌入金属有机骨架;④表面修饰的方法。
1.1 非手性物质在晶体生长过程中自组装形成具有螺旋结构的手性MOF
在没有纯手性物质存在的情况下,通过非手性物质在结晶过程中自组装形成具有螺旋结构的MOF,这种螺旋结构的存在能使三维网络结构产生手性,如包含3折和4折螺旋的(10,3)结构。采用廉价的非手性试剂合成出手性金属有机骨架具有很大的优势,因为它避免了采用通常比较昂贵的手性试剂(一般要采用比较繁琐的步骤才能得到)。然而正如Lin等[12]评述的那样,“那些宣称采用这种自识别方法组装成的同手性金属有机骨架实际上几乎全部都是外消旋(即非手性)的”。采用非手性物质自组装得到手性MOF例子较少,其大部分倾向于得到外消旋的配位聚合物[13]。
Martin等[16]用Co(NO3)2·6H2O、含氧二(安息香酸)(H2oba)和二(4-吡啶基甲基)哌嗪(bpmp)为原料,在120℃下反应72h,得到手性3D配位聚合物[Co3(oba)3(bpmp)2]n。该配合物是由相同类型但不同手性的无限螺旋链通过{Co3O2}簇连接成具有8-连接的单节4451767拓扑型的连环3D框架结构。Liu等[17]在水热条件下,以Zn(CH3COO)2·2H2O、苯三甲酸(H3tma)、p,p′-二胺-二苯基-甲烷(ddm)为原料合成了一种3D手性配位聚合物Zn[Htma][ddm]。该配位聚合物具有P212121手性空间群,中心Zn原子通过具有△构型的刚性三角构造与配体tma及柔性分子ddm桥联,形成具有3D螺旋金刚石框架的结构。Fan等[18]采用非手性的1,4-萘二甲酸(H2L)和CdCl2在DMF溶剂中160℃水热合成了3D手性金属有机骨架,结构简式为[Cd(μ6-L)(DMF)]n。该配位聚合物具有P212121手性空间群,且具有两种不同类型的1D Cd-O-Cd螺旋链,两种Cd-O-Cd螺旋链沿b轴相互交织在一起,然后沿a轴和c轴与1,4-萘二甲酸联接形成3D框架结构。
Shen等[19]采用非手性配体2,2-联吡啶-5,5-二羧酸(H2BPDC)、甲酸与Zn(NO3)2·6H2O水热合成了手性MOF,结构简式为 Zn3(BPDC)2(O2CH)2·2DEF。该MOF具有手性空间群P41212,BPDC-Zn2+螺旋链的存在使其具有手性孔道。Sun等[20]采用3,5-吡啶二甲酸(H2PDC)、MnCl2、N,N-二甲基甲酰胺(DMF)和乙二醇在55℃加热3天合成了3D手性金属有机骨架JUC-58,结构简式为Mn(3,5-PDC)(H2O)·(glycol)。每个Mn(II)分别与4个配体H2PDC上的羧基氧原子、另一配体H2PDC上的氮原子以及水分子上的氧原子配位,形成双锥体结构。每个羧基上的两个氧原子分别与两个不同的Mn(II)原子配位,形成Mn-O-C螺旋链,这些螺旋链通过配体上的N原子连接形成3D手性结构。
1.2 利用手性化合物诱导合成具有手性特征的手性MOF
利用手性化合物诱导合成手性MOF是合成手性MOF的有效方法之一。但是手性化合物一般价格昂贵,合成成本较高。Rosseinsky等[21-22]利用纯光学活性试剂1,2-丙二醇(1,2-pd)作为模板剂,均三苯甲酸(H3btc)、嘧啶(py)/3-甲基吡啶(3-pic)作为配体与六水合硝酸镍合成了两种不同的手性MOF,结构简式分别为Ni3btc2(py)6(1,2-pd)3- [(1,2-pd)11(H2O)8]和 Ni3btc2(3-pic)6(1,2-pd)3- [(1,2-pd)9(H2O)6]。它们的结构中含有两种互相贯通的(10,3)螺旋结构。Morris等[23]利用含有手性阴离子的离子液体1-丁基-3-甲基咪唑L-天冬氨酸盐作为反应介质,得到了手性SIMOF-1,结构简式为(BMIm)2[Ni(btc-H)2(H2O)2],每个Ni与4个苯三甲酸和两个水分子配位饱和形成八配位的结构,且具有手性空间群P41212,对称的41轴产生了螺旋状的假四面体单元,从而使其具有了手性。
1.3 通过旋光纯的手性有机基团与金属离子配位将手性成分嵌入MOF
从手性配体出发来合成目标手性产物是一种最直接、最有效的合成手性MOF的方法。常用的手性配体主要包括一些手性氨基酸及其衍生物[24-26]、手性金属有机化合物[27-28]、多肽类手性化合物[29-30]。手性氨基酸及其衍生物同时包含氨基和羧基,为含N、O配体,具有良好的配位能力,在配合物中形成超分子结构时,分别充当形成氢键的受体和给予体。但是通常氨基酸配体与含羧基的有机阴离子或含氮杂环有机中性配体等辅助配体混合使用,配体上的氧原子和氮原子为氢键的形成提供了必要条件,且第二配体大多选择含有苯环结构的,如对苯二甲酸[4]、4,4-联吡啶[25]等,苯环结构的存在使得π-π相互作用成为可能,对稳定和拓展晶体结构起重要作用。手性大分子有机化合物或手性金属有机化合物一般合成步骤比较复杂,价格昂贵。
1.3.1 手性氨基酸及其衍生物作为手性配体
D-樟脑酸是构筑手性MOF较常用的旋光纯物质之一,这是由于它的结构中含有一个五元环,其形成的手性MOF结构稳定性好。Liang及其同事[24]利用D-樟脑酸和四(4-吡啶氧甲基)甲烷(TPOM)分别与Zn(NO3)2·6H2O和Cd(NO3)2·4H2O在水/乙醇溶剂中水热合成了两种纯手性MOF,结构简式为M2(TPOM)(D-cam)2(H2O)2(M=Zn、Cd)。单晶X射线衍射表明其均具有P21212的手性空间群,为正交晶系。D-樟脑酸与M金属离子首先形成螺旋1D手性链[M(D-cam)]n,然后螺旋1D手性链[M(D-cam)]n与波状2D [M2(TPOM)]n通过金属离子相互交织形成3D网络结构。且根据手性材料的分类表,两种MOF均具有C1A2类型的结构,即它们是由1D手性链与2D非手性链连接而成。
为此我们对山羊颗粒TMR进行了研究,在对制粒工艺完成初步试验后,设计了山羊颗粒TMR的适口性及采食行为的观察试验,以检验在生产中应用的可行性[1],并为以颗粒TMR为基础的饲养管理新方式提供参考;同时确定其进行可量产的成套设备和生产线开发的必要[2]。在按设计要求成功加工出山羊全混合日粮颗粒料后,为进一步优化工艺,进行了几种不同加工方式的颗粒饲料贮存观察。可为肉羊全混合颗粒饲料的开发应用提供参考。
Zhang等[25]对比研究了对映纯的D-樟脑酸和外消旋的D,L-樟脑酸对形成的MOF结构的影响。他们分别采用D-樟脑酸/外消旋的D,L-樟脑酸与4,4′-联吡啶(4,4′-bpy)/1,2-二(4-吡啶基)乙烯(bpe)作为混合配体,分别与Cu2+、Zn2+和Cd2+水热合成了6种配位聚合物,结构简式分别为[Cu2(D-cam)2(4,4′-bpy)]n(1c)、[Cu2(DL-cam)2(4,4′-bpy)]n(1a)、[Zn2(D-cam)2(4,4′-bpy)]n(2c)、[Zn2(DL-cam)2(4,4′-bpy)]n(2a)、[Cd4(D-cam)4(bpe)3]n(3c)、[Cd2(DL-cam)2(bpe)3/2]n(3a)。研究发现采用对映纯D-樟脑酸合成的1c、2c和3c均为手性MOF,而采用外消旋的D,L-樟脑酸合成的1a,2a和3a为外消旋的MOF。1c与1a、2c与2a具有相近的晶胞参数和晶体结构,且金属离子Cu2+、Zn2+与D-樟脑酸的配位模式相同。3c与3a的晶胞参数和晶体结构明显不同,Cd2+与D-樟脑酸的配位模式不同,且分别有两种不同的配位模式。Huang等[31]采用D-樟脑酸的衍生物成功地合成出两种具有双螺旋结构的MOF,其之所以具有独特的互相交织的双螺旋结构,是由于其独特的手性螺旋配体1,2,2-三甲基-3-(4-吡啶基)氨基甲酰基环戊烷羧酸[1,2,2-trimethyl-3-(pyridin-4-ylcarbamoyl) cyclopentanecarboxylic acid,L]。L配体分别与Zn(NO3)2·6H2O和Cd(NO3)2·4H2O 120℃、2天水热合成了MOF,结构简式分别为ZnL2(H2O)2和CdL2(H2O)2。
L-苹果酸含有两个羧基桥连基团和一个羟基,可与金属中心产生多种多样的连接方式,并能显示出双齿、三齿、四齿、五齿等多种配位方式,因此L-苹果酸是构筑手性MOF的常用旋光纯有机配体之一。Beghidja等[32]采用手性桥联配体苹果酸(mal)与Cd2+反应制备得到了新型手性3D MOF [Cd((S)-mal)]。Cd2+处于变形的五角双锥构型,分别与苹果酸的7个氧原子配位,其中6个氧原子属于羧基氧,1个属于α-羟基氧。每个中心Cd2+通过这两种氧原子与4个相邻的Cd2+相连。每个苹果酸与5个Cd2+配位。Cd2+间由苹果酸连成具有无限结构的手性3D结构。Areg等[33]采用硝酸锰、L-苹果酸、琥珀酸、4,4-联吡啶、碳酸钠、乙醇、水在120℃水热反应5天制备得到了两种手性MOF,结构简式分别为[Mn2(L-ma)0.4(suc)1.6(4,4′-bipy)2(H2O)2]·4,4′-bipy (1) 和 [Mn(L-ma)(4,4′-bipy)(H2O)]·3H2O (2)。配合物 1和2是采用相同的混合物制备得到的,但是L-苹果酸与Mn2+的配位模式不同,在配合物2中L-苹果酸中的羧基氧和羟基氧分别参与配位,而在配合物1中只有羧基氧参与配位。Nagaraja等[34]采用L-苹 果 酸(L-mal)与4,4-偶 氮 联 吡 啶(4,4-bisazobipyridine,azpy)或1,2-双(4-吡啶基)乙烯[1,2-bis(4-pyridyl)ethylene,bpee]分别与Co(Ⅱ)或Ni(Ⅱ)反应制备得到了4种同构型的3D多孔金属有机配位聚合物,结构简式为{[M(L-mal)(azpy)0.5]·2H2O} [M=Co(1)、Ni(2)]和{[M(L-mal)(bpee)0.5]·H2O} [M=Co(3)、Ni(4)]。4种配位聚合物均为正交晶系,且具有手性空间群P21212。
Dybtsev等[4]利 用Zn(NO3)2·6H2O、L-乳酸(L-H2lac)和1,4-对苯二甲酸(H2bdc)以摩尔比2∶1∶1水热合成了一种棒状的纯手性MOF材料,结构简式为[Zn2(bdc)(L-lac)(dmf)]·(DMF) (1化合物·DMF)。Zn2+与L-乳酸沿a轴形成1D手性链,然后沿b、c轴分别与对苯二甲酸相互连接形成3D网络结构。作者课题组采用L-乳酸作为手性配体,2-氨基对苯二甲酸作为非手性配体,与六水合硝酸锌水热制备了手性金属有机骨架CUP-1,结构简式为Zn2(atpt)(l-lac)(HCOO),其中甲酸是由DMF水解得到的。CUP-1具有正交晶系,手性空间群P2(1)2(1)2(1)[35]。
Ingleson等[26]利用L-或D-天冬氨酸和4,4′-联吡啶通过溶剂热方法合成了手性MOF,结构简式为Cu(asp)bpe0.5(H2O)。韩国浦项工科Kim研究组[36]以光学纯的有机建筑模块手性酒石酸衍生物[D-型或L-型,图1(a)]与锌离子反应得到了具有二维结构的多孔材料D-POST-1或L-POST-1,结构简式为[Zn3(μ3-O)1-H6]·2H3O·12H2O。单晶X射线衍射数据表明其层间距为15.47Å(1Å=0.1mm),层与层之间通过范德华作用力沿着c轴方向堆积,形成边长为13.4 Å的等边三角形1D手性孔道。Xiong等[37]利用手性配体S-羧甲基-N-(p-甲苯磺酰基)-L-巯基丙氨酸(Ts-cmc)、非手性配体4,4′-bipy与锰盐反应,合成了3D手性配位聚合物[Mn(C12H13NO6S2)(C10H8N2)(H2O)]n。该配合物中Mn2+处于扭曲的八面体构型。每个Ts-cmc配体具有μ3-连接节点,连接相邻的Mn2+形成二维框架结构,然后通过4,4′-bipy连接形成手性3D结构。最近,Chen及其同事[38]采用均苯三甲酰三(L-丙氨酸) (L-TMTAH3)、4,4-联吡啶(4,4-bpy)、甲醇和水室温合成了一种手性MOF,结构简式为[Zn(L-TMTA)2(4,4-bpy)4]·24H2O。其结构中存在两种不同的ZnII离子,一种ZnII离子与不同配体L-TMTA3-上的4个氧原子和2个4,4-bpy配体上的氮原子配位形成扭曲的八面体结构。另一种ZnII离子与不同配体L-TMTA3-的2个羧基上的3个氧原子和3个4,4-bpy配体上的3个氮原子配位形成扭曲的八面体结构。每个L-TMTA3-与4个ZnII离子配位。
1.3.2 手性大分子有机化合物

图1 反应物质的结构式
Wu等[5]利用手性大分子有机配体(R)-6,6′-二 氯-2,2′-二羟基-1,1′-双萘-4,4′-双吡啶和CdCl2在MeOH/DMF混合溶剂中慢扩散合成了一种纯手性 金属-有机多孔状超分子,结构简式为[Cd3Cl6L3]·4DMF·6MeOH·3H2O (1)。Rossin等[39]采用(R,R)-四氢噻唑-2,4-二羧酸(H2L-RR)和COCl2·6H2O水热合成了手性MOF,结构简式为[Co(L-RR)(H2O)·H2O]∞。该MOF具有手性空间群P41,且为四方晶系。Lestari等[40]采用(S)-5,5-双(4-羧基苯基)-2,2-双(二苯基磷酰基)-1,1-联萘(H2L)、Pb(NO3)2、NaOH和DMF水热合成了3D双金属手性MOF,结构简式为{[Na2Pb2(L)3(H2O)(dmf)4]·9dmf}n。Mantion 研究组[29-30]首先采用多肽类手性化合物(图2)作为有机配体合成手性MOF。他们采用低聚肽手性化合物Z-(L-Val)2-L-Glu(OH)-OH[图2(b)]与Ca2+和Cu2+自组装制备了两种新型手性MOF[30]。
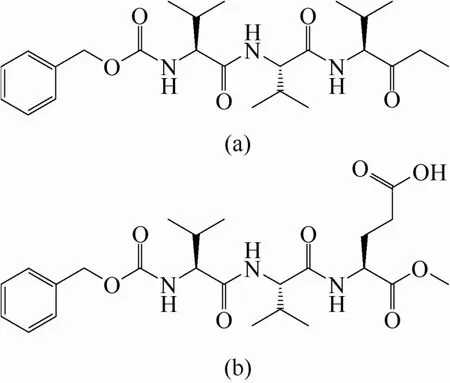
图2 多肽手性化合物结构式
1.3.3 手性金属有机化合物
Cho等[27]利用手性金属锰有机配体(R,R)-(-)-1,2-环己基二胺-N,N′-双(3-叔丁基-5-(4-吡啶基)亚水杨基)MnIIICl [L,图1(b)]、非手性配体4,4-联苯二甲酸 (H2bpdc) 和Zn2+水热法合成了一种手性MOF材料,结构简式为Zn2(bpdc)2L·10DMF·8H2O。Cho等[28]报道了几种由双邻苯二酚手性金属锰络合物{[bis(catechol)salen]MnIII,图1(c)}和几种二价或三价的金属离子配位聚合形成的手性配位聚合物poly(M-1)[M=CuⅡ、CrⅢ、MnⅡ、FeⅢ、CoⅡ、NiⅡ、ZnⅡ、CdⅡ、MgⅡ,图1(d)]。
1.4 表面修饰的方法
表面修饰的方法又叫后合成共价修饰法,即用手性的有机基团通过键连或表面锚装的方法修饰一些已知的MOF材料。这种策略作为在非手性金属有机骨架上创立催化活性位的方法早有报道[41-42]。然而直到2009年,Banerjee等[43]首次报道采用后合成共价修饰法构筑手性金属有机骨架,他们以MIL-101为基体,采用手性有机配体(S)-N-(吡啶-3-基)吡咯烷-2-甲酰胺[(S)-N-(pyridine-3-yl)-pyrrolidine-2-carboxamide](L1) 和(S)-N-(吡 啶-4-基)吡 咯 烷-2-甲 酰 胺[(S)-N-(pyridine-4-yl)-pyrrolidine-2-carboxamide](L2)进行后合成修饰,利用L1或L2中的N与MIL-101中配位不饱和的Cr(Ⅲ)离子配位形成手性金属有机骨架,并用于醛酮的Aldol反应。Demuynck等[44-46]采用三氟甲磺酸酐和硫磺酸对MIL-101(Cr)进行后合成共价修饰得到硫酸盐化的S-MIL-101(Cr),然后采用手性二元胺[如2-(S)-氨基-3-苯丙酸二辛胺]对S-MIL-101(Cr)进行后合成共价修饰构筑了手性的diamine/S-MIL-101(Cr)催化剂。该催化剂对羟醛缩合不对称催化反应具有较高的对映体选择性。
如前所述,在创立手性MOF的4种方法中,通过旋光纯的手性有机配体与金属离子配位将手性成分嵌入MOF的方法,是在合成MOF时至少选择一种手性有机分子作为配体,手性配体与金属离子自组装制备手性MOF的方法,该方法比较简单,是比较常用的方法之一,目前合成的手性MOF大多采用这种方法;缺点是大多与其他含氮、含羧基有机配体混合使用。利用表面修饰法得到手性MOF是行之有效的方法之一,其缺点是大多数MOF的金属离子大多被配体配位饱和,不存在配位不饱和的金属离子,且具有特定有机官能团的有机配体是有限的,因而采用表面修饰法制备纯手性MOF只能针对某些特定的MOF(如IRMOF-3、MIL-101)。利用非手性配体和手性模板剂合成纯手性MOF的方法比较困难,因此这两种方法也受到一定的限制。
2 手性MOF在不对称催化反应中的应用
近年来,手性金属有机骨架化合物的合成发展迅速,并取得了一些喜人的成果,一些MOF材料已经具有手性拆分和不对称催化性能[4-6,27-28]。目前MOF材料已经成功地应用于加成[5]、环氧化[27,47]、醇分解[26]、酯交换[36]以及羟醛缩合[43]等不对称催化反应中。但是,手性MOF作为催化材料催化不对称反应的例子相对较少,直到2000年,Kim研究组[36]才在Nature上首次报道了手性金属有机骨架D-POST-1或L-POST-1具有对映体选择分离和催化功能。吡啶基团暴露在D-POST-1或L-POST-1孔道内部,因此,不但可以对其进行化学改性,且由于氮原子的存在,氮原子有多余的电子,可以提供电子,作为Lewis碱催化活性位。D-POST-1或L-POST-1催化2,4-二硝基苯乙酸酯与过量1-苯基-2-丙醇消旋混合物反应,得到相应的S或R酯对映体选择性(ee%)为8%。此值虽小,但这是首次发现MOF材料具有不对称催化性质,因而具有相当重要的意义,为以后进一步提高对映选择性奠定了基础。
随后,Wu等[5]报道了手性配体(R)-6,6′-二氯-2,2′-二羟基-1,1′-双萘-4,4′-双吡啶与Cd2+合成的一种手性金属有机多孔状超分子在催化二乙基锌[Zn(Et)2]与1-萘甲醛加成反应时,1-萘甲醛几乎完全反应(转化率大于99%),对映体(R)-1-(1-萘基)丙醇的选择性(ee%)达到93%,远高于Kim研究组报道的D-POST-1或L-POST-1在催化酯基交换反应中的对映体过量值(ee%)8%[36]。且该催化剂对芳香醛具有较宽的适用范围,在催化ZnEt2与其他芳香醛(如4-氯苯甲醛、3-溴苯甲醛)的加成不对称催化反应中产率一般都在90%以上,对映体过量值也在80%左右,对映体选择性与均相催化剂BINOL/Ti(OPr)4相差不大,对反应物具有尺寸效应,见图3。
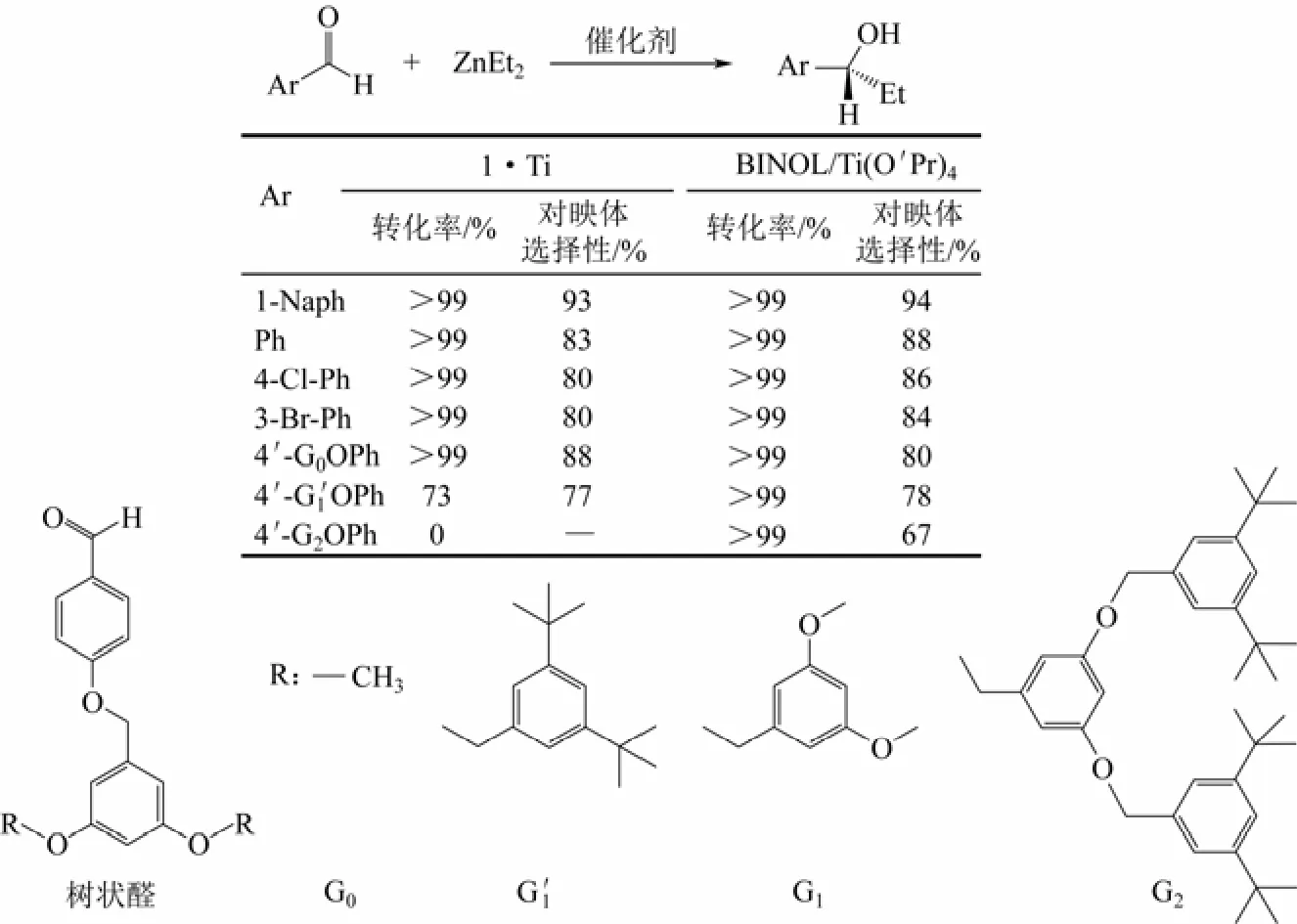
图3 芳香醛和二乙基锌加成反应
Dybtsev等[4]利用Zn(NO3)2·6H2O ,L-乳酸(L-H2lac)和1,4-对苯二甲酸(H2bdc)水热合成了手性MOF材料,结构简式为[Zn2(bdc)(L-lac)(dmf)]·(DMF)(1化合物·DMF)。其对硫醚氧化为硫氧化物的反应具有较好的催化活性和选择性,见图4。当氧化剂为尿素过氧化氢复合物(UHP)时,具有较小取代基团的2a和2b(图4)的转化率分别为64%和58%,选择性分别为92%和83%,而具有较大取代基团的2c和2d的转化率只有7%和3%。用90%H2O2代替UHP时,2a和2b同样得到了100%、85%的转化率,87%、100%的选择性。且催化剂可以重复利用至少30次而没有氧化物选择性的降低。在氧化反应后,加入一定量的1化合物,反应产物如(S)-3a以20%(ee%)被优先吸附到孔道中,而R对映体被留在液相中,从而实现了对映异构体选择性分离。
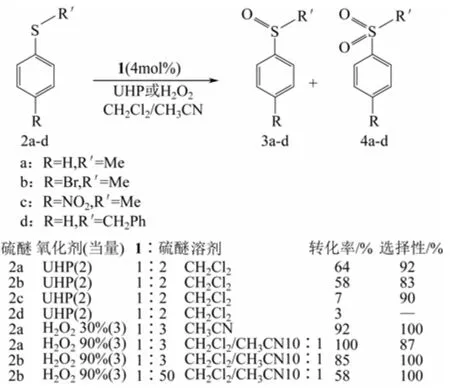
图4 硫醚的氧化反应
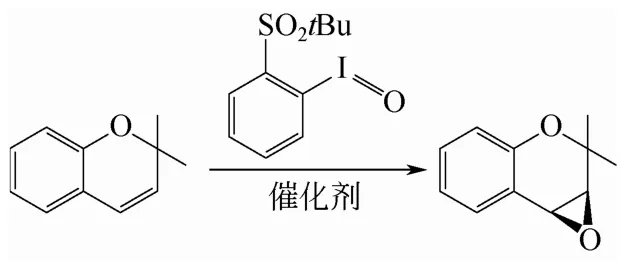
图5 2,2-二甲基-2H-色烯的不对称环氧化反应
Cho等[27]利用手性配体(R,R)-(-)-1,2-环己基二胺-N,N′-双(3-叔丁基-5-(4-吡啶基)亚水杨基)MnIIICl,4,4-联苯二甲酸(H2bpdc)和Zn2+水热法合成了一种MOF材料,结构简式为Zn2(bpdc)2L·10DMF·8H2O。其 在 催 化2,2-二 甲 基-2H-色烯(2,2-dimethyl-2H-chromene)的不对称环氧化反应中(氧化剂为2-叔丁基磺酰基亚碘酰苯)(图5),对映体选择性(ee%)为82%,仅比均相催化剂Lee%稍低(88%),且容易分离,可以循环使用;而均相催化剂L在最初几分钟活性损失很大,在反应3.4h后活性几乎没有,且分离困难。Cho 等[28]报道了双邻苯二酚手性金属锰络合物(图1中c)和几种二价或三价的金属离子配位聚合形成的手性配位聚合物,poly(M-1)(M=CuⅡ、CrⅢ、MnⅡ、FeⅢ、CoⅡ、NiⅡ、ZnⅡ、CdⅡ、MgⅡ,图1中d)在 2,2-二甲基-2H-色烯环氧化反应中具有较好的催化作用。氧化剂为2-叔丁基磺酰基亚碘酰苯,olefin/oxidant/catalyst的摩尔比为100∶150∶1时,poly(Cu-1)作为催化剂时产率为79%,对映体选择性(ee%)为76%,且可以循环使用10次,产率稍有降低,从79%降到70%,对映体选择性几乎没有改变(ee%75%~76%)。在相同的反应条件下,poly(Cr-1)、poly(Mn-1)、poly(Cd-1)和poly(Mg-1)作为催化剂时的产率均在70%~89%,poly(Co-1)和poly(Zn-1)的产率分别为49%和66%,poly(Fe-1)和poly(Ni-1)的产率最低只有22%和31%。但是poly(Cr-1)、poly(Cd-1)、poly(Mg-1)和poly(Zn-1)的对映体选择性较高为76%;poly(Mn-1)和poly(Co-1)的对映体选择性次之,分别为60%和52%;poly(Fe-1)和poly(Ni-1)的对映体选择性只有48%和20%。
Ingleson等[26]利用L-或D-天冬氨酸和4,4′-联吡啶通过溶剂热方法合成了手性MOF,他们利用无水HCl对该手性MOF的羧酸配体质子化作用得到了一种含有B酸活性位的材料,结构简式分别为Cu(L-asp)bpe0.5(HCl)(H2O)或Cu(D-asp)bpe0.5(HCl)- (H2O)。这两种手性MOF在25℃催化顺式-2,3-环氧丁烷的甲醇分解反应时(图6),得到的(R,R)和(S,S)的产物的总产率分别为59%和65%,ee%(对映体选择性)分别为10%和-6%,TOF为4.8和4.7;在0℃反应时产率分别为30%和32%,ee%分别为17%和-13%,TOF分别为2.6和2.7。其ee%远高于均相催化剂H2SO4,采用H2SO4作为催化剂时,尽管产率高达100%,但ee%仅为2%(25℃)。
Candu等[47]采用二聚Cr(Ⅲ)手性化合物对金属有机骨架[Cu2(mand)2(hmt)]后合成共价修饰得到手性的dimeric CrIII-salen/[Cu2(mand)2(hmt)]作为催化剂,研究了其在反式-肉桂酸甲酯环氧化制备(2R,3S)-苯基环氧丙酸甲酯的反应中的催化活性和对映体选择。采用5%(摩尔分数) dimeric CrIII-salen/[Cu2(mand)2(hmt)]催化剂,室温下反应72h后,氧化剂为H2O2时,反式-肉桂酸甲酯的转化率为9.1%,(2R,3S)-苯基环氧丙酸甲酯ee%为53.9%。当氧化剂为N-甲基吗啉-N-氧化物(NMO) 时,转化率和ee%分别为48.5%和81.6%,且环氧化物的选择性为100%。
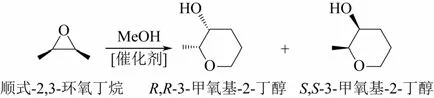
图6 顺式-2,3-环氧丁烷的甲醇分解反应
Bogaerts等[50]将Mn(Ⅲ)手性化合物包裹在MIL-101(Al)孔道中,制备得到了多相催化剂Mn-salen@MIL101(Al)。其在二氢化萘环氧化反应中,二氢化萘的转化率为69%,对映体选择性(ee%)为70%,其转化率略低于相同用量的均相Mn(Ⅲ)手性化合物(82%),但是其对映体选择性与均相催化剂的基本相同,高达70%。且该催化剂至少可以循环使用4次,其TON和ee%基本保持不变。
Demuynck等[44]采用后合成共价修饰制备了手性diamine/S-MIL-101(Cr)催化剂,并发现其对直链酮和芳香醛的不对称羟醛缩合反应具有较高的催化活性和对映体选择性(产率49%~94%,ee%61%~98%)。diamine/S-MIL-101(Cr)的对映体选择性与MIL-101的一致,但是其催化活性明显高于MIL-101的(2-丁酮和4-三氟甲基苯甲醛在室温下反应20h后产率分别为53%和18%,ee%均为96%)。他们还研究了diamine/S-MIL-101(Cr)对反应底物的适用范围,发现其对于2-丁酮、3-戊酮和羟丙酮均具有较好的催化活性和较高的对映体选择性,但是其对于丙酮和2-戊酮的催化活性和对映体选择性较低。diamine/S-MIL-101催化剂可以循环使用4次,其催化活性略有降低,但是其ee%基本保持不变(前4次循环使用时,羟丙酮和4-三氟甲基苯甲醛在室温下分别反应20h、24h、26h和30h,产率分别为97%、94%、93%和90%,ee%分别为95%、93%、94%和92%)。
Banerjee等[43]利用手性有机配体(S)-N-[吡啶-3-基)吡咯烷-2-甲酰胺] (L1) 和(S)-N-[吡啶-3-基)吡咯烷-2-甲酰胺] (L2) 对MIL-101进行表面修饰,得到了两种手性金属有机多孔材料CMIL-1和CMIL-2。CMILs在催化不同的芳香醛和酮发生的羟醛缩合不对称反应中羟酮的产率在60%~90%,R-异构体的对映体选择性在55%~80%(图7)。该催化剂的活性与均相催化剂的相差不大,对映体选择性却高于均相催化剂L1或L2(ee%为21%~66%)。比如,CMIL-1在室温催化对硝基苯甲醛和丙酮反应的产率为66%,对映体选择性为69%。在相同的反应条件下均相催化剂L1催化该反应,产率为58%,对映体选择性仅为29%。而没有改性的MIL-101反应5天后转化率仅有10%(0,ee%)。CMIL-1至少可以重复使用3次,产率和对映体选择性基本保持不变。
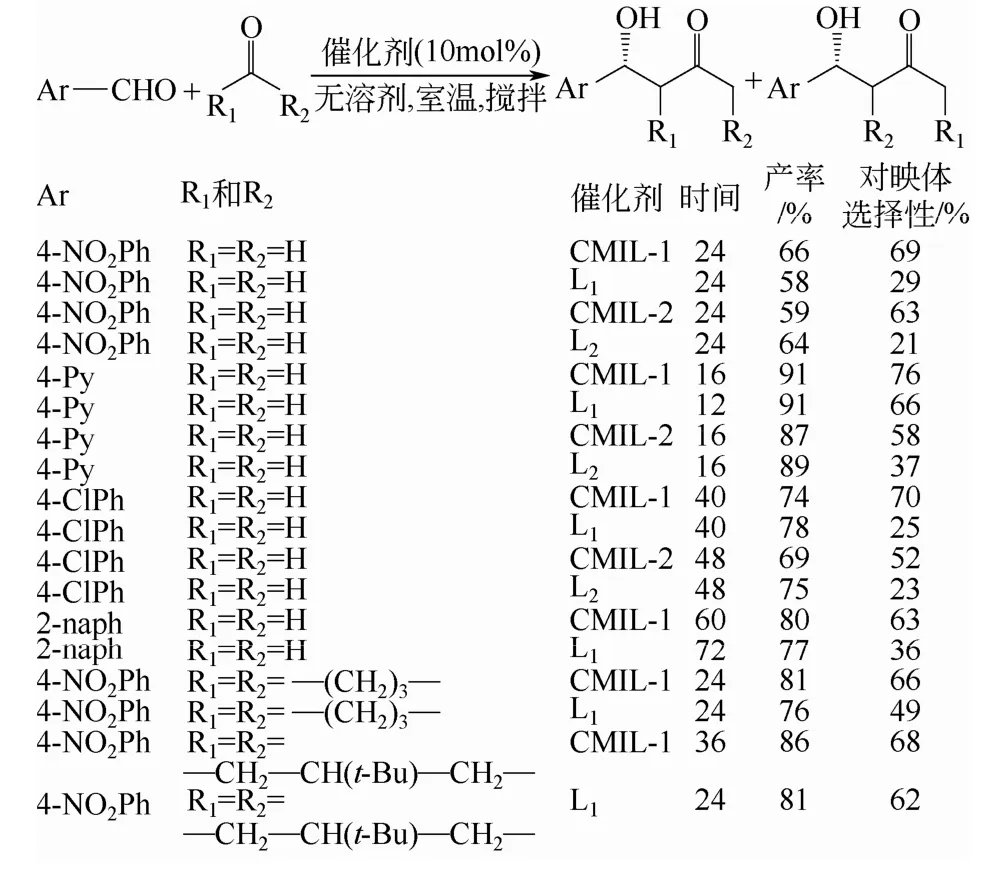
图7 芳香醛和酮的羟醛缩合不对称反应
最近,Zhu等[49]采用L-AMP对Zn-DPPG后合 成共价修饰得到了手性金属有机骨架 Zn-Fun1,其在4-硝基苯甲醛和环己酮羟醛缩合不对称反应中具有较高的反应活性(产率85%)和对映体选择性(ee%为77%)。其对映体选择性远远高于均相催化剂L-AMP的对映体选择性(ee%为26%)。Zhu等[49]还采用L-AMP和D-AMP对Zn-DPYI后合成共价修饰得到了两种手性MOF Zn-MOF1和Zn-MOF2,两种催化剂在醛和酮的羟醛缩合不对称反应中具有相似的催化活性和对映体选择性,且得到完全相反的对映体(在25℃反应7d,4-硝基苯甲醛和环己酮Aldol反应产率分别为75%和76%,ee%分别为70%和-73%)。
3 结 语
综上所述,手性MOF合成及其在不对称催化反应中的应用已成为研究的一个新热点。尽管经过近几年的努力,目前手性MOF在不对称催化领域的研究已取得较大进展,然而手性MOF 在不对称催化领域中应用研究还处于初级发展阶段。这主要体现在以下几方面。
(1)到目前为止,对手性MOF 的研究仍主要集中在新型手性MOF的设计、制备和表征上,向功能化扩展是今后发展的必然趋势。
(2)合成手性MOF的金属离子大多采用Zn、Cd、Ni等过渡金属离子,采用镧系金属离子(La、Ce、Pr、Nd、Sm、Eu、Tb、Dy)及碱性金属离子(Mg、Ca)的较少。
(3)手性MOF所特有的结构特性与可能具备的独特不对称催化性能还需要更多的例子来证明,目前手性MOF大部分只对特定的反应,甚至只对特定的反应底物有效,反应类型比较单一,还需要对反应的多样化进行探索。
(4)合成制备手性MOF的方法还需要大力探索和开发,尤其是在合成制备具有不对称催化活性的手性MOF的方法有待进一步的研究。
(5)多数手性MOF存在转化数较低、对映体选择性较低、重复使用性能差等不足。
因此,如何设计合成高效、新型的手性MOF催化剂,探讨配体和催化剂设计的规律,解决手性MOF催化剂的选择性和稳定性,发展系列重要的不对称反应的手性MOF催化剂是未来研究需要考虑的一个重要方向。
[1] Wang Z,Chen G,Ding K L. Self-supported catalysts [J].Chem. Rev.,2009,109(2):322-359.
[2] Fan Q H,Ding K L. Enantioselective catalysis with structurally tunable immobilized catalysts[J].Top.Organometal.Chem.,2011,36:207-246.
[3] 邱惠斌. 手性介孔材料的合成、形成机理及性能[M]. 上海:上海交通大学,2010.
[4] Dybtsev D N,Nuzhdin A L,Chun H,et al. A homochiral metal-organic material with permanent porosity,enantioselective sorption properties,and catalytic activity[J].Angew.Chem.Int.Ed.,2006,45(6):916-920.
[5] Wu C D,Hu A,Zhang L,et al. A homochiral prous metal-organic framework for highly enantioselective heterogeneous asymmetric catalysis[J].J.Am.Chem.Soc.,2005,127:8940-8941.
[6] Mueller U,Schubert M,Teich F J. Metal-organic frameworks-prospective industrial application[J].Mater.Chem.,2006,16:626-636.
[7] 陈恒,陈绍云,苑兴洲,等. 咪唑衍生物辅助合成金属有机骨架MIL-101及CO2吸附性能[J]. 化工进展,2014,33(7):1808-1815.
[8] 肖冰心,刘杰,王双,等.离子热合成金属-有机骨架材料最新研究进展[J]. 化工进展,2014,33(9),2363-2371.
[9] Leong W L,Vittal J J. One-dimensional coordination polymers:Complexity and diversity in structures,properties,and applications[J].Chem.Rev.,2011,111:688-764.
[10] 张所瀛,刘红,刘朋飞,等. 金属有机骨架材料在CO2/CH4吸附分离中的研究进展[J]. 化工学报,2014,65(5):1563-1570.
[l1] 刘丽丽,张鑫,徐春明. MOF基上创立活性位的方法及其催化应用[J]. 化学进展,2010,22(11):2089-2098.
[12] Ma L Q,Abney C,Lin W B. Enantioselective catalysis with homochiral metal-organic frameworks[J].Chem.Soc.Rev.,2009,38(5):1248-1256.
[13] 汪海明,王正,丁奎岭. 手性自负载催化剂研究新进展[J]. 化学进展,2010,22(7):1471-1481.
[14] Falkowski J M,Liu S,Lin W B. Metal-organic frameworks as single-site solid catalysts for asymmetric reactions[J].Isr. J. Chem.,2012,52(7):591-603.
[15] Yoon M,Srirambalaji R,Kim K. Homochiral metal-organic frameworks for asymmetric heterogeneous catalysis[J].Chem.Rev.,2012,112(2):1196-1231.
[16] Martin D P,Staples R J,LaDuca R L. A chiral self-catenated dual-ligand coordination polymer constructed from three distinct interwoven helical motifs interconnected by one-dimensional chains[J].Inorg.Chem.,2008,47(21):9754-9756.
[17] Liu L,Huang S P,Yang G D,et al. Zn[Htma][ddm]: An interesting three-dimensional chiral nonlinear optical-active zinc-trimesate framework[J].Cryst.Growth Des.,2010,10(2):930-936.
[18] Fan Z,Wu X X,Ding B,et al. Hydrothermal synthesis and characterization of a novel 3D chiral inorganic-organic hybrid cadmium(Ⅱ) framework with achiral 1,4-naphthalenedicarboxylic acid ligand[J].Synth.React.Inorg.M,2014,44:371-375.
[19] Shen L J,Gray D,Masel R I,et al. Synthesis and characterization of a zinc metal-organic framework with chiral nano-pores[J].Cryst. Eng. Comm.,2012,14:5145-5147.
[20] Sun F X,Zhu G S. Solvent-directed synthesis of chiral and non-centrosymmetric metal-organic frameworks based on pyridine-3,5-dicarboxylate[J].Inorg.Chem.Commun.,2013,38:115-18.
[21] Kepert C J,Prior T J,Rosseinsky M J. A versatile family of interconvertible microporous chiral molecular frameworks:The first example of ligand control of network chirality[J].J.Am.Chem.Soc.,2000,122(21):5158-5168.
[22] Bradshaw D,Prior T J,Cussen E J,et al. Permanent microporosity and enantioselective sorption in a chiral open framework[J].J.Am.Chem.Soc.,2004,126(19):6106-6114.
[23] Lin Z J,Slawin A M Z,Morris R E. Chiral induction in the ionothermal synthesis of a coordination polymer[J].J.Am.Chem.Soc.,2007,129(16):4880-4881.
[24] Liang L L,Ren S B,Zhang J,et al. Two thermostable three-dimensional homochiral metal-organic polymers with quartz topology[J].Cryst.Growth Des.,2010,10(3):1307-1311.
[25] Zhang J,Yao Y G,Bu X H. Comparative study of homochiral and racemic chiral metal-organic frameworks built from camphoric acid[J].Chem.Mater.,2007,19(21):5083-5089.
[26] Ingleson M J,Barrio J P,Bacsa J,et al. Generation of a solid Brøsted acid site in a chiral framework[J].Chem.Commun.,2008,11:1287-1289.
[27] Cho S H,Ma B Q,Nguyen S T,et al. A metal-organic framework material that functions as an enantioselective catalyst for olefin epoxidation[J].Chem.Commun.,2006,2563-2565.
[28] Cho S H,Gadzikwa T,Afshari M,et al.[Bis(catechol)salen]MnIIIcoordination polymers as support-free heterogeneous asymmetric catalysts for epoxidation[J].Eur.J.Inorg.Chem.,2007,31:4863-4867.
[29] Mantion A,Taubert A. TiO2sphere-tube-fiber transition induced by oligovaline concentration variation[J].Macromol.Biosci.,2007,7(2):208-217.
[30] Mantion A,Massüger L,Rabu P,et al. Metal-peptide frameworks (MPFs):“Bioinspired”metal organic frameworks[J].J.Am.Chem.Soc.,2008,130:2517-2526.
[31] Huang Q,Yu J C,Gao J K,et al. Two chiral nonlinear optical coordination networks based on interwoven two-dimensional square grids of double helices[J].Cryst.Growth Des.,2010,10(12):5291-5296.
[32] Beghidja A,Beghidja C,Rogez G,et al. Synthesis,crystal structure and spectroscopic properties of a new chiral cadmium (Ⅱ) malate[J].Inorg.Chem.Commun.,2008,11(9):1088-1090.
[33] Areg A,Zhang J,Bu X H. Cooperative self-assembly of chiral L-malate and achiral succinate in the formation of a three-dimensional homochiral framework[J].Inorg.Chem.,2008,47(19):8607-8609.
[34] Nagaraja C M,Haldar R,Maji T K,et al. Chiral porous metal-organic frameworks of Co(Ⅱ) and Ni(Ⅱ):Synthesis,structure,magnetic properties,and CO2uptake[J].Cryst.Growth Des.,2012,12:975-981.
[35] Liu L L,Zhang X,Rang S M,et al. Catalysis by metal-organic frameworks:Proline and gold functionalized MOFs for the aldol and three-component coupling reactions[J].RSC Adv.,2014,4:13093-13107.
[36] Seo J S,Whang D,Lee H,et al. A homochiral metal-organic porous material for enantioselective separation and catalysis[J].Nature,2000,404(6781):982-986.
[37] Xiong W,Su Y,Chen Z,et al. A three-dimensional homochiral metal-organic framework constructed from manganese(Ⅱ) withS-carboxymethyl-N-(p-tosyl)-L-cysteine and 4,4′-bipyridine[J].Acta Cryst.,2009,C65(2):m56-m58.
[38] Chen Z L,Zhang C B,Liu X L,et al. Synthesis,structure,and properties of a chiral zinc(Ⅱ) metal-organic framework featuring linear trinuclear Secondary Building Blocks[J].Aust.J.Chem.,2012,65:1662-1666.
[39] Rossin A,Credico B D,Giambastiani G,et al. Synthesis,characterization and CO2uptake of a chiral Co(Ⅱ) metal-organic framework containing a thiazolidine-based spacer[J].J.Mater.Chem.,2012,22:10335-10344.
[40] Lestari W W,Lönnecke P,Streit H C,et al. Synthesis,structure and luminescence properties of a three-dimensional heterobimetallic chiral metal-organic framework based on sodium(Ⅰ),lead(Ⅱ) and (S)-5,5-Bis(4-carboxyphenyl)-2,2-bis(diphenylphosphinoyl)-1,1-bina phthyl as linker[J].Eur.J.Inorg.Chem.,2014,1775-1782.
[41] Tanabe K K,Wang Z Q,Cohen S M. Systematic functionalization of a metal-organic frameworkviaa postsynthetic modification approach[J].J.Am.Chem.Soc.,2008,130:8508-8517.
[42] Garibay S J,Wang Z Q,Tanabe K K,et al. Postsynthetic modification:A versatile approach toward multifunctional metal-organic framework[J].Inorg.Chem.,2009,48:7341-7349.
[43] Banerjee M,Das S,Yoon M,et al. Postsynthetic modification switches an achiral framework to catalytically active homochiral metal-organic porous materials[J].J.Am.Chem.Soc.,2009,131(22):7524-7525.
[44] Demuynck A L W,Goesten M G,Ramos-Fernandez E V,et al. Induced chirality in a metal-organic framework by postsynthetic modification for highly selective asymmetric Aldol reactions[J].Chem. Cat. Chem.,2014,6:2211-2214.
[45] Goesten M G,Juan-Alcañiz J,Ramos-Fernandez E V,et al. Sulfation of metal-organic frameworks:Opportunities for acid catalysis and proton conductivity[J].J.Catal.,2011,281(1):177- 187.
[46] Juan-Alcañiz J,Gielisse R,Lago A B,et al. Towards acid MOFs-catalytic performance of sulfonic acid functionalized architectures[J].Catal.Sci.Technol.,2013,3(9):2311-2318.
[47] Candu N,Tudorache M,Florea M,et al. Postsynthetic modification of a metal-organic framework (MOF) structure for enantioselective catalytic epoxidation[J].Chem. Plus. Chem.,2013,78:443-450.
[48] Zhu W T,He C,Wu P Y,et al. “Click” post-synthetic modification of metal-organic frameworks with chiral functional adduct for heterogeneous asymmetric catalysis[J].Dalton Trans.,2012,41:3072-3077.
[49] Zhu W T,He C,Wu X,et al. “Click” post-synthetic modification of metal-organic frameworks for asymmetric aldol catalysis[J].Inorganic Chem.Commun.,2014,39:83-85.
[50] Bogaerts T,Deyne A V Y,Liu Y Y,et al. Mn-salen@MIL101(Al):A heterogeneous,enantioselective catalyst synthesized using a“bottle around the ship”approach[J].Chem.Commun.,2013,49:8021-8023.
