Ce改性对Y型分子筛酸性及其催化转化性能的调变机制
2015-08-15秦玉才高雄厚张海涛莫周胜初春雨张晓彤宋丽娟辽宁石油化工大学辽宁省石油化工催化科学与技术重点实验室辽宁抚顺00中国石油天然气股份有限公司石油化工研究院兰州化工研究中心兰州70060中国石油大学华东化学工程学院山东青岛66555
张 畅 秦玉才 高雄厚 张海涛 莫周胜初春雨 张晓彤 宋丽娟,,*(辽宁石油化工大学,辽宁省石油化工催化科学与技术重点实验室,辽宁抚顺00;中国石油天然气股份有限公司石油化工研究院兰州化工研究中心,兰州70060;中国石油大学(华东),化学工程学院,山东青岛66555)
Ce改性对Y型分子筛酸性及其催化转化性能的调变机制
张畅1秦玉才1高雄厚2,*张海涛2莫周胜3初春雨1张晓彤1宋丽娟1,3,*
(1辽宁石油化工大学,辽宁省石油化工催化科学与技术重点实验室,辽宁抚顺113001;2中国石油天然气股份有限公司石油化工研究院兰州化工研究中心,兰州730060;3中国石油大学(华东),化学工程学院,山东青岛266555)
采用液相离子交换(LPIE)法制备了不同离子交换度的CeY分子筛.运用电感耦合等离子发射光谱(ICPAES)、X射线衍射(XRD)、N2吸附等温线和氨气程序升温脱附(NH3-TPD)等方法对其进行表征,采用原位傅里叶变换红外(in situ FTIR)光谱技术分别以吡啶和噻吩作为探针分子研究了Ce改性对Y型分子筛酸性能和催化转化性能的影响规律.结果表明,Ce离子改性不改变Y型分子筛晶体的基本骨架,但改变其精细结构.分子筛改性过程中Ce物种优先定位于方钠石(SOD)笼,随着稀土离子含量增大,逐渐出现在超笼中.Ce离子交换过程中产生一定量的Brönsted(B)酸中心,且其量与强度随着Ce含量的增大均呈现先增加后平稳的趋势.同时,Ce离子交换产生与非骨架铝物种和铈物种有关的两种强度不同的Lewis(L)酸中心,且两者均随着Ce含量的增大而增大.噻吩吸附红外光谱表明,由于Ce离子改性产生的强B酸中心可导致噻吩在室温条件下即可发生质子化反应,质子化的噻吩分子可进一步发生低聚反应.而稀土物种与B酸中心的协同作用有利于低聚反应的发生.
原位傅里叶变换红外光谱;CeY;酸性;噻吩;催化转化
www.whxb.pku.edu.cn
1 引言
稀土改性Y型分子筛(REY)是流化催化裂化(FCC)催化剂的主活性组分,自其被用于催化裂化反应以来,关于REY分子筛中稀土离子的落位与形态,以及其对分子筛骨架结构、水热稳定性、酸性能和催化性能的影响等方面的研究一直没有间断过.1-16普遍认为其优异的催化裂化性能取决于稀土改性赋予了Y型分子筛一些独特的物理和化学性能.改性稀土离子进入Y型分子筛SOD笼SIʹ位,与骨架O原子相互作用,增强了分子筛骨架结构和水热稳定性;7-9同时,稀土改性还可以调变分子筛的酸性,10-14但其调变机制至今仍存在很多争议.分子筛改性过程中,由于稀土水合离子的离解作用使吸附水发生极化,从而产生Brǒnsted(B)酸中心,同时分子筛骨架脱铝产生的非骨架铝等物种和稀土物种本身会形成分子筛的Lewis(L)酸中心.而有关改性分子筛中B酸和L酸中心的酸性与稀土物种的含量、形态、落位等性质的关联及其相互影响方面的研究还不够细致系统.
关于改性Y型分子筛中稀土物种的形态结构方面已有很多报道.大多学者认为稀土物种是以[RE(OH)]2+或[RE(OH)2]+,或两种羟基结构共存的形式存在于Y分子筛中,15,16然而关于此稀土物种的酸性质及其在催化转化反应中具体扮演的角色等更深层次方面的工作还鲜有报道.因此,系统开展有关分子筛改性过程中酸中心的产生机理,稀土极化水分子产生B酸中心的能力和所产生L酸中心的种类和强度等性质,以及稀土在分子筛中的含量、存在状态与存在环境对酸中心和催化转化性能影响规律方面的工作,对于研发催化剂新材料具有重要的指导意义.
本文采用液相离子交换法制备了不同Ce含量(质量分数,w)的Y型分子筛,运用ICP-AES、XRD和N2吸附等表征手段对其进行了详细表征.利用吡啶吸附FTIR(Py-FTIR)技术探讨Ce改性对Y型分子筛酸性的调变机制.以噻吩催化转化为模型反应,采用In situ FTIR技术系统考察稀土离子改性调变酸性后,各类酸中心在催化转化反应过程中具体扮演的角色,为合成出高效催化转化性能的催化剂提供理论依据.
2 实验部分
2.1原料与试剂
NaY和NH4Y原粉(硅铝比2.55,南开大学催化剂厂);吡啶、噻吩(分析纯,百灵威化学试剂有限公司);硝酸铈(分析纯,国药集团化学试剂有限公司).
2.2分子筛样品的制备
通过传统的离子交换法制备得到不同Ce含量的CeY分子筛,17分别记作L-CeY-1、L-CeY-2和LCeY-3.L-CeY-1是通过NaY分子筛和0.02 mol·L-1的Ce(NO3)3溶液经一次交换一次焙烧制得,焙烧前样品记作L-CeY-1(hydrated);L-CeY-2是通过NaY分子筛和0.06 mol·L-1的Ce(NO3)3溶液经一次交换一次焙烧制得;L-CeY-3是通过NaY分子筛和0.06 mol·L-1的Ce(NO3)3溶液经二次交换二次焙烧制得;NH4Y分子筛直接焙烧得到HY分子筛;NH4Y分子筛和0.06 mol·L-1的Ce(NO3)3溶液经一次交换一次焙烧制得CeHY分子筛.
2.3样品的表证
采用美国热电(Thermo Elemental)iCAP 6300型电感耦合等离子发射光谱仪(ICP-AES)测定离子交换度;样品的物相分析采用日本理学株式会社D/ MAX-RB型X射线衍射仪(XRD),Cu靶Kα射线,入射波长为0.154 nm,管电压30 kV,管电流100 mA,5°-60°范围扫描,扫描速率8(°)·min-1,连续扫描;样品的比表面积和孔体积在美国麦克(Micromeritics)公司生产的ASAP 2020型物理吸附仪上测定,催化剂样品在623 K下抽真空预处理10 h,液氮冷却至77 K,进行低温N2吸/脱附实验,并利用BET (Brunauer-Emmett-Teller)法计算比表面积,HK(Horvath-Kawazoe)法计算微孔的孔容和孔径分布,BJH(Barrett-Joyner-Halenda)法计算介孔的孔容和孔径分布.分子筛酸强度和酸量分布测试在美国麦克(Micromeritics)公司生产的AutoChem II 2920型全自动程序升温化学吸附仪上完成.
2.4酸性及催化转化行为的测定
二者的测定在美国Perkin-Elmer公司Frontier型傅里叶变换红外(FTIR)光谱仪上进行.样品经研细压制成自支撑薄片(10-12 mg·cm-2),装入CaF2窗片的石英红外吸收池里,程序升温加热至673 K恒温,真空(10-3Pa)活化4 h,然后降至室温,对样品进行扫描.在298 K下吸附探针分子30 min,之后当以吡啶为探针分子时,分别在423和673 K抽真空脱附30 min,用红外光谱仪扫描谱图.分子筛在423 K脱附后的特征峰面积定义为总酸量,在673 K脱附后的峰面积定义为强酸量,两者之差为弱酸量.以噻吩为探针分子时,分别在298、373、473和573 K下抽真空脱附30 min,降至室温后,用红外光谱仪对样品进行扫描.红外光谱仪扫描波数范围均为4000-1200 cm-1,扫描次数为32次,分辨率为4 cm-1.

表1 样品的Na+交换度(B)和孔结构Table 1 Ion exchange extent of Na+(BNa+)and the physical properties of the zeolite samples used
3 结果与讨论
3.1改性分子筛的结构性能考察
改性分子筛的Na离子交换度计算结果见表1,图1为NaY、L-CeY-1(hydrated)、L-CeY-1、L-CeY-2 和L-CeY-3五种分子筛的XRD谱图,插图为部分区域放大图像.由图1可见,L-CeY-1(hydrated)(b′)、LCeY-1(b)、L-CeY-2(c)及L-CeY-3(d)分子筛的(220)面、(311)面和(331)面的衍射强度明显减弱,而其它衍射面强度未见明显变化,表明液相Ce离子交换过程中分子筛的基本晶体结构仍保持完整,仅对晶体的精细结构造成一定影响.18,19L-CeY-1分子筛脱水前(b′)后(b)(222)面衍射峰与(311)面衍射强度变化表明,离子交换后稀土离子首先进入超笼,焙烧后再迁移至SOD笼.20-23相关研究也已有证明,稀土物种易定位于Y分子筛中SOD笼的SI′位.7,19,24由LCeY-1(b)、L-CeY-2(c)和L-CeY-3(d)的XRD谱图可见,随着Ce离子含量的增加,一部分Ce物种出现在超笼中,从而导致(222)面衍射峰的出现,且随着Ce含量的增加此衍射峰逐渐增强(如图1(c,d)所示),表明随着离子交换度的增加,超笼中Ce物种逐渐增多.25,26
图2为NaY、L-CeY-1、L-CeY-2和L-CeY-3四种分子筛的N2吸附-脱附等温线,由此计算出的分子筛织构性质列于表1.结果表明,随着Ce离子含量的增加,L-CeY分子筛吸附和脱附分支间出现了较窄的滞后环.由图2中插图可见,高稀土含量时,分子筛中产生了孔径分布较宽的介孔,说明分子筛在改性过程中发生了脱铝或脱硅过程,导致了分子筛中产生部分骨架缺陷位,进而导致如表1所示的比表面积及孔容的下降.
3.2分子筛的酸性能调变
图3为分子筛NaY(a)、L-CeY-1(b)、L-CeY-2(c)和L-CeY-3(d)的NH3-TPD谱图.与改性前NaY相比,改性后三种分子筛弱酸中心均向高温区移动,且总量相对减少.随着稀土含量的增大,三种分子筛的中强酸中心量先增大后减小,强酸中心量略有增加.
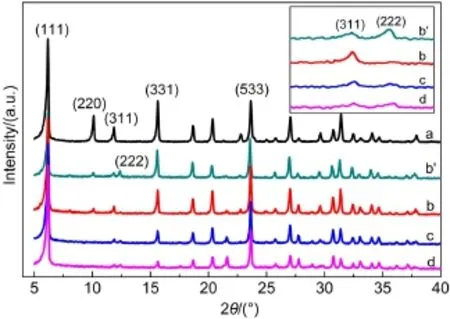
图1 分子筛的XRD谱图Fig.1 XRD patterns of the zeolites
图4为NaY(A),L-CeY-1(B),L-CeY-2(C)和LCeY-3(D)四种分子筛的Py-FTIR谱图.从图中可见,NaY分子筛与吡啶作用,423 K脱附后只产生了归属于弱L酸位的1442 cm-1特征吸收峰,15同时归属于本底非骨架铝羟基的特征吸收峰(3694 cm-1)(见图4A曲线a)消失.27673 K脱附后,1442 cm-1特征吸收峰消失,非骨架铝羟基特征吸收峰又恢复,结合NH3-TPD结果,表明NaY分子筛中的Na+和非骨架铝羟基均为弱的L酸位.
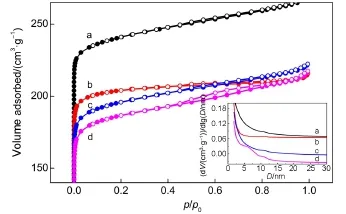
图2 分子筛的N2吸附-脱附等温线和BJH法介孔分布图(插图)Fig.2 N2adsorption-desorption isotherms and BJH mesopore size distributions(inset)of the zeolites(a)NaY;(b)L-CeY-1;(c)L-CeY-2;(d)L-CeY-3

图3 分子筛的NH3-TPD谱图Fig.3 NH3-TPD spectra of the zeolites(a)NaY;(b)L-CeY-1;(c)L-CeY-2;(d)L-CeY-3
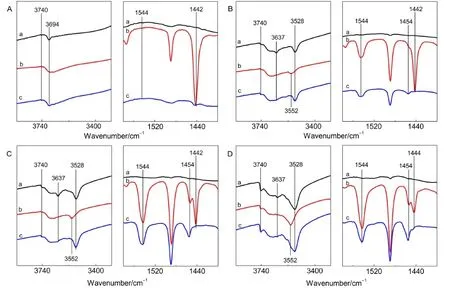
图4 NaY(A),L-CeY-1(B),L-CeY-2(C)和L-CeY-3(D)分子筛的Py-FTIR谱图Fig.4 Py-FTIR spectra of NaY(A),L-CeY-1(B),L-CeY-2(C)and L-CeY-3(D)zeolites(a)background;(b)pyridine desorption at 423 K;(c)pyridine desorption at 673 K
与NaY分子筛不同,稀土改性后的三种分子筛吸附吡啶后均产生了归属于B酸位的1544 cm-1特征吸收峰,且由423和673 K不同脱附温度下的结果表明,Ce改性后分子筛均产生了强弱两种B酸中心.从分子筛羟基变化情况看,Ce改性后分子筛产生了分别归属于超笼中的硅铝桥羟基(3637 cm-1)和与稀土物种有关的羟基(3528 cm-1),28-32其与吡啶分子作用后,前者低温脱附时完全消失,673 K脱附后有微弱恢复,而后者低温时减弱并发生蓝移,高温下完全恢复,表明三种分子筛超笼中桥羟基绝大部分为强B酸中心,与稀土物种有关的羟基为弱B酸中心,其具体形成机制如图5所示.
分子筛改性过程中,Ce离子极化水解:Ce3++ H2O→[Ce(OH)]2++H+,Ce3++2H2O→[Ce(OH)2]++2H+,产生的H+与超笼中硅铝桥氧原子相结合,形成3637 cm-1硅铝桥羟基(图5中a物种),同时产生了[Ce(OH)]2+(图5中b物种)或[Ce(OH)2]+(图5中c物种)等与Ce离子直接相连的羟基结构(3528 cm-1).图6为L-CeY-1(hydrated)分子筛焙烧过程中羟基变化的原位FTIR谱图,从图中可以看出,随着焙烧温度的升高,3637和3528 cm-1处两种羟基逐渐出现,这一结果与我们上面所阐述的分子筛经Ce改性后酸性中心的形成机制正相吻合.而且从图4中三种改性分子筛673 K谱图可见,1544 cm-1吸收峰面积先增大后不再增加,表明分子筛中强B酸中心密度随着稀土含量增大呈现先增大后平缓趋势.Ce物种通过对超笼中桥羟基电子对的吸引作用增强其B酸中心强度,随着Ce离子含量的增大,极化作用增强.33但由于有些位于SOD笼中的Na+不易被交换掉,当Ce含量达到一定程度后不再交换Na+,而是相互聚集形成Ce的二或三聚物等,致其对羟基的极化作用不再增强,或是稀土过多时,由于平均每个RE3+不能极化出最多的H+,稀土离子甚至进入超笼,故而使分子筛的强B酸中心量不再增加.24

图5 分子筛焙烧脱水过程中酸性位的产生机制示意图Fig.5 Schematic of mechanism of acid sites generated in the dehydration process of zeolites(a)cerium;(b)hydrone;(c)sodium;(d)alumina bridge hydroxy;(e)[Ce(OH)]2+;(f)[Ce(OH)2]+;(g)Si-OH;(h)non-framework aluminum species;(i)defects in an aluminum species
从图4还可以观察到,三种改性分子筛吸附吡啶后均出现1454 cm-1吸收峰,其归属于与非骨架铝和缺陷位铝等物种有关的L酸中心,34表明分子筛改性会导致骨架脱铝和脱硅,产生的非骨架铝(图5中e物种)和缺陷位铝(图5中f物种)成为了分子筛的L酸中心.图6中随焙烧温度的升高,分子筛中非骨架铝羟基(3694 cm-1)逐渐增多的现象也更好地证实分子筛改性过程中伴随着骨架脱铝.
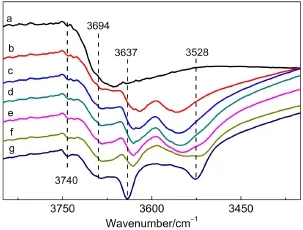
图6 L-CeY-1(hydrated)分子筛脱水过程中羟基伸缩区域变化情况的FTIR谱图Fig.6 FTIR spectra of OH stretching region changes of L-CeY-1(hydrated)during calcination(a)298 K;(b)423 K;(c)473 K;(d)523 K;(e)573 K;(f)623 K;(g)673 K
另外,L-CeY-3分子筛吸附吡啶后在1444 cm-1波数处出现另一新的吸收峰,其归属为吡啶与Ce物种作用产生的,15,28,29673 K脱附后该峰完全消失,表明分子筛中存在一种新的弱L酸中心.结合423 K下与稀土有关的3528 cm-1处羟基峰蓝移(从3528至3552 cm-1)并于673 K脱附后完全恢复的现象,可得此种新的弱L酸中心即为与此蓝移部分羟基直接相连的阳离子.吡啶作用在Ce离子上后,由于吡啶的供电作用使与Ce离子相连接的羟基氧上的孤对电子向H离子偏移,使羟基能量增加进而导致蓝移现象的产生,Sousa-Aguiar等31,32的实验结果也证实了这一点.而当Ce含量很小时,分子筛与吡啶作用后产生1442 cm-1吸收峰,且峰型很尖锐,其仍归属于分子筛中还存在的未交换完全的Na+与吡啶作用后产生的,当然引入的稀土离子也有少许贡献,如(图4(B,C)).图7为HY和CeHY两种分子筛的FTIR谱图,从图7(A)可见,HY分子筛只存在一种与非骨架铝物种有关的L酸中心(1454 cm-1),而在制备过程中排除了Na+影响的CeHY分子筛中存在两种L酸中心(1454和1444 cm-1)(图7B),对比可得,1444 cm-1处吸收峰确为吡啶与稀土物种作用产生,再次证明了此种L酸中心与稀土物种有关,且随着离子交换度的增大,与Ce物种相关的弱L酸中心逐渐增多.
3.3分子筛的催化转化性能调变
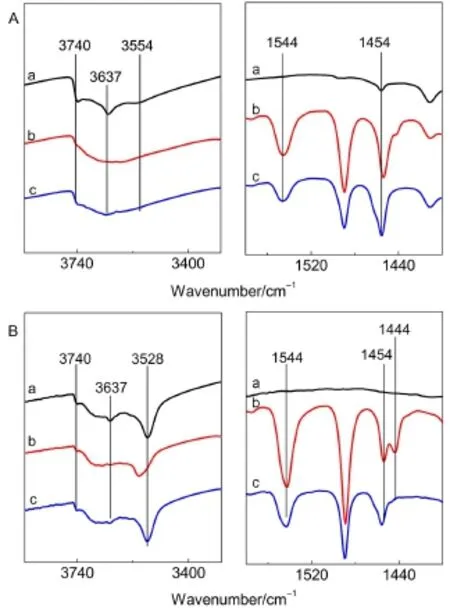
图7 HY(A)和CeHY(B)分子筛的Py-FTIR谱图Fig.7 Py-FTIR spectra of the HY(A)and CeHY(B)zeolites(a)background;(b)pyridine desorption at 423 K;(c)pyridine desorption at 673 K
图8为噻吩在分子筛NaY(A)、L-CeY-1(B)、LCeY-2(C)和L-CeY-3(D)吸附的FTIR谱图.从图中可知,室温下噻吩在NaY分子筛上吸附后出现3106 cm-1(归属于吸附态噻吩分子的不饱和CH伸缩振动)和1396 cm-1(归属于吸附态噻吩的CH面外弯曲振动)两个吸收峰,并且非骨架铝羟基吸收峰(3694 cm-1)完全消失,吸附的噻吩在373 K条件下可完全脱附,3694 cm-1处羟基吸收峰也逐渐恢复,表明噻吩分子分别与NaY分子筛中钠离子和非骨架铝羟基发生π电子相互作用和氢键作用,机理如图9 (a)所示.
从图8(B,C,D)可知,室温条件下,噻吩在三种L-CeY分子筛上吸附后在3106和1396 cm-1波数处也出现特征吸收峰,但两吸收峰强度均随着Ce离子交换度的增加逐渐减弱,表明随着Ce离子含量的增大,分子筛上吸附态噻吩的量逐渐减少,该结果也证实Ce离子交换后剩余的Na+主要分布于SOD笼中噻吩分子较难接近的位置.此外,红外谱图中还出现了1504、1449、1439和1408 cm-1四个新的吸收峰.吸收峰1449和1439 cm-1归属于*CH2―S―或者*CH2―(CH=CH2)两种结构中的CH2的弯曲振动.其中,1449 cm-1归属于质子化后产生的CH2弯曲振动,而1439 cm-1则归属于噻吩低聚物中的饱和CH2的弯曲振动.另外,1504 cm-1归属于噻吩低聚物环的伸缩振动吸收峰,而1408 cm-1则归属于噻吩的环伸缩振动.3,28,29,35-37同时,噻吩吸附后三种分子筛的超笼中桥羟基(3637 cm-1)完全消失,直至高温下也未恢复完全.结合酸性表征结果表明噻吩吸附在此三种分子筛上后在强B酸中心作用下发生了质子化反应和低聚反应.
对比噻吩吸附FTIR谱图可见,三种不同离子交换度的CeY分子筛上1504、1449和1439 cm-1三个新的特征吸收峰的强度明显不同,其中噻吩在LCeY-1分子筛上吸附后仅1449 cm-1处的吸收峰稍强,而1504和1439 cm-1波数处特征峰很弱,说明噻吩仅发生了较明显的质子化反应,而几乎不发生低聚反应.而室温下噻吩吸附在L-CeY-2和L-CeY-3两种分子筛上后,在1504、1449和1439 cm-1处均产生了明显的吸收峰,表明室温下噻吩分子在此两种分子筛上即可发生剧烈的质子化反应和低聚反应.而从373 K脱附时L-CeY-3分子筛上1504和1439 cm-1处吸收峰的增强程度明显强于L-CeY-2分子筛.可见,L-CeY-3分子筛对噻吩的低聚反应能力明显强于L-CeY-2.另外,473 K下,噻吩在L-CeY-2和L-CeY-3两种分子筛上还均在1415 cm-1处产生一较宽的吸收峰,可归属于稠环大分子产物的特征峰,随着脱附温度的升高该峰逐渐消失,表明生成的噻吩低聚物逐渐聚合成稠环大分子,并于高温下逐渐分解.28结合四种分子筛的酸性表征结果及以上分析结果可得,室温条件下噻吩在分子筛强B酸中心作用下即可发生质子化反应,如图8(B,C,D)所示;质子化的噻吩分子可在稀土物种(1444 cm-1)和非骨架铝有关的L酸中心(1454 cm-1)的促进作用下进一步发生低聚反应,如图8(C,D)所示.而两者含量越大,越利于增加吸附在分子筛上噻吩的密集程度,进而利于质子化的噻吩分子发生低聚反应,如图8 (D)所示.噻吩在L-CeY分子筛上质子化和低聚反应机理如图9(b)所示.
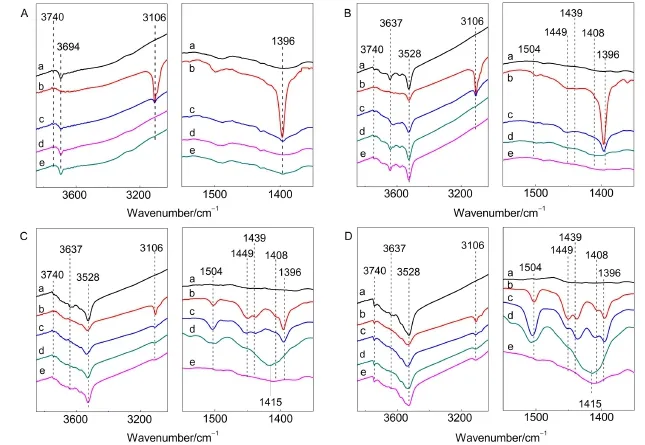
图8 噻吩在NaY(A),L-CeY-1(B),L-CeY-2(C)和L-CeY-3(D)分子筛上吸附的FTIR谱图Fig.8 FTIR spectra of thiophene adsorbed on the NaY(A),L-CeY-1(B),L-CeY-2(C)and L-CeY-3(D)zeolites(a)background;(b)thiophene desorption at 298 K;(c)thiophene desorption at 373 K;(d)thiophene desorption at 473 K;(e)thiophene desorption at 573 K

图9 噻吩吸附在分子筛上的转化机理Fig.9 Conversion mechanisms of thiophene on the Y zeolites(a)NaY;(b)L-CeY
图10为噻吩在HY(A)和CeHY(B)分子筛上吸附的FTIR谱图,从图中可看出,噻吩在HY和CeHY分子筛上均可发生质子化反应和低聚反应,而对比可见,噻吩质子化产物在CeHY分子筛上进一步发生低聚反应的能力明显强于HY.并结合图7中二者的酸性结果,HY中仅含有与非骨架铝物种有关的强L酸中心(1454 cm-1),而CeHY中同时存在与非骨架铝物种有关的强L酸中心和与稀土物种有关的弱L酸中心(1444 cm-1),且前者的强L酸中心密度明显大于后者,可有力地证实出与稀土物种有关的弱L酸中心(1444 cm-1)在促进噻吩质子化产物向低聚物的转化的过程中起主要作用.有关烃类分子的转化行为及非骨架铝物种在催化转化过程中所起作用等问题的研究正在进行中.
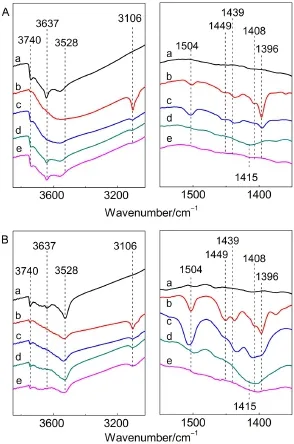
图10 噻吩在HY(A)和CeHY(B)分子筛上吸附的FTIR谱图Fig.10 FTIR spectra of thiophene adsorbed on the HY(A)and CeHY(B)zeolites(a)background;(b)thiophene desorption at 298 K;(c)thiophene desorption at 373 K;(d)thiophene desorption at 473 K;(e)thiophene desorption at 573 K
4 结论
分子筛改性过程中,Ce离子对水分子的极化作用产生B酸中心,且其量与强度均随着稀土含量的增大呈先增加后平稳的趋势.Ce离子交换过程中分子筛发生脱铝脱硅,产生与非骨架铝等物种有关的强L酸中心;生成的类似[Ce(OH)]2+或[Ce(OH)2]+等铈物种是弱L酸中心.室温条件下,噻吩即可在强B酸中心作用下发生质子化反应,质子化产物可在L酸中心作用下进一步发生低聚反应,而稀土物种与B酸的协同作用更有利于促进质子化噻吩向低聚物的转化.
References
(2)Wang,D.;Liu,T.;Cai,J.P.;Zhai,J.N.;Zhang,Z.D.Applied Chemical Industry 2014,43(1),165.[王栋,刘涛,蔡军平,翟佳宁,张忠东.应用化工,2014,43(1),165.]
(3)Shi,Y.C.;Zhang,W.;Zhang,H.X.;Tian,F.P.;Jia,C.Y.;Chen,Y.Y.Fuel Process.Technol.2013,110,24.doi:10.1016/j. fuproc.2013.01.008
(4)Pang,X.M.;Gu,Y.S.;Zhang,L.;Sun,S.H.;Gao,X.H. Petrochemical Technology 2004,33(S0),1478.[庞新梅,谷育生,张莉,孙书红,高雄厚.石油化工,2004,33(增刊),1478.]
(5)Duan,L.H.;Gao,X.H.;Meng,X.H.;Zhang,H.T.;Wang,Q.;Qin,Y.C.;Zhang,X.T.;Song,L.J.J.Phys.Chem.C 2012,116 (49),25748.doi:10.1021/jp303040m
(6)Lappas,A.A.;Valla,J.A.;Vasalos,I.A.;Kuehler,C.;Francis,J.;Connor,P.O′.;Guddec,N.J.Appl.Catal.A:Gen.2004,262 (1),31.doi:10.1016/j.apcata.2003.11.014
(7)Yu,S.Q.;Tian,H.P.;Dai,Z.Y.;Long,J.Chin.J.Catal.2010,31(10),1263.[于善青,田辉平,代振宇,龙军.催化学报,2010,31(10),1263.]
(8)Thomas,B.;Sugunan,S.Microporous Mesoporous Mat.2006,95,55.
(9)Fan,M.G.;Li,B.;Zhang,F.Y.;Li,W.L.;Xing,J.M.;Liu,Z. L.Acta Phys.-Chim.Sin.2009,25(3),495.[范闽光,李斌,张飞跃,李望良,邢建民,刘自力.物理化学学报,2009,25(3),495.]doi:10.3866/PKU.WHXB20090316
(10)Yu,S.Q.;Tian,H.P.;Zhu,Y.X.;Dai,Z.Y.;Long,J.Acta Phys.-Chim.Sin.2011,27(11),2528.[于善青,田辉平,朱玉霞,代振宇,龙军.物理化学学报,2011,27(11),2528.]doi:10.3866/PKU.WHXB20111101
(11)Huang,F.L.;Li,X.F.;Wang,L.G.Chemical Engineering of Oil and Gas 2008,37(2),122.[黄风林,李小锋,王立功.石油与天然气化工,2008,37(2),122.]
(12)Martins,G.V.A.;Berlier,G.;Bisio,C.;Coluccia,S.;Pastore,H.O.;Marchese,L.J.Phys.Chem.C 2008,112,7193.doi:10.1021/jp710613q
(13)Duan,H.C.;Li,X.L.;Zhang,H.T.;Tan,Z.G.;Gao,X.H. Chemical Engineering&Equipment 2012,10,5.[段宏昌,李雪礼,张海涛,谭争国,高雄厚.化学工程与装备,2012,10,5.]
(14)Garcia,F.A.C.;Araújo,D.R.,Silva,J.C.M.;de Macedo,J.L.;Ghesti,G.F.;Dias,S.C.L.;Diasa,J.A.;Filhoc,G.N.R. J.Braz.Chem.Soc.2011,22(10),1894.doi:10.1590/S0103-50532011001000010
(16)Xue,Z.Y.;Zhu,L.M.;Dai,L.S.Acta Phys.-Chim.Sin.1990,6 (2),183.[薛志元,朱雷明,戴林森.物理化学学报,1990,6 (2),183.]doi:10.3866/PKU.WHXB19900211
(17)Jin,L.L.;Li,X.Q.;Wang,H.G.;Ju,X.F.;Liu,L.M.;Song,L. J.Industrial Catalysis 2008,16(10),31.[靳玲玲,李秀奇,王洪国,鞠秀芳,刘黎明,宋丽娟.工业催化,2008,16(10),31.]
(18)Du,X.H.;Gao,X.H.;Zhang,H.T.;Li,X.L.;Liu,P.S.Catal. Commun.2013,35,17.doi:10.1016/j.catcom.2013.02.010
(19)Nery,J.G.;Giotto,M.V.;Mascarenhas,Y.M.;Cardoso,D.;Zotin,F.M.Z.;Sousa-Aguiar,E.F.Microporous Mesoporous Mat.2000,41,281.doi:10.1016/S1387-1811(00)00304-8
(22)Du,X.H.;Zhang,H.T.;Li,X.L.;Tan,Z.G.;Liu,H.H.;Gao,X.H.Chin.J.Catal.2013,34,1599.doi:10.1016/S1872-2067 (11)60622-6
(23)Wang,N.N.;Wang,Y.;Cheng,H.F.;Fu,M.;Tao,Z.;Wu,W.Z. J.Porous.Mater.2013,20,1371.doi:10.1007/s10934-013-9723-1
(24)Li,B.;Li,S.J.;Li,N.;Liu,C.H.;Gao,X.H.;Pang,X.M. Chin.J.Catal.2005,26(4),301.[李斌,李士杰,李能,刘丛华,高雄厚,庞新梅.催化学报,2005,26(4),301.]
(25)Li,X.W.;Yu,L.Q.;Liu,X.Y.Chin.J.Catal.1982,3(1),34.[李宣文,余励勤,刘兴云.催化学报,1982,3(1),34.]
(26)Zhang,T.;Zhou,L.P.;Tian,H.P.Petroleum Processing and Petrochemical Technology 2011,42(4),40.[张探,周灵萍,田辉平.石油炼制与化工,2011,42(4),40.]
(27)Datka,J.;Sulikowski,B.;Gil,B.J.Phys.Chem.1996,100(27),11242.doi:10.1021/jp951523+
(28)Qin,Y.C.;Gao,X.H.;Pei,T.T.;Zheng,L.G.;Wang,L.;Mo,Z.S.;Song,L.J.Journal of Fuel Chemistry Technology 2013,41(7),889.[秦玉才,高雄厚,裴婷婷,郑兰歌,王琳,莫周胜,宋丽娟.燃料化学学报,2013,41(7),889.]
(29)Zhang,X.T.;Yu,W.G.;Qin,Y.C.;Dong,S.W.;Pei,T.T.;Wang,L.;Song,L.J.Acta Phys.-Chim.Sin.2013,29(6),1273.[张晓彤,于文广,秦玉才,董世伟,裴婷婷,王琳,宋丽娟.物理化学学报,2013,29(6),1273.]doi:10.3866/PKU. WHXB201303183
(30)Wang,N.N.;Wang,Y.;Cheng,H.F.;Tao,Z.;Wang,J.;Wu,W. Z.RSC Adv.2013,3,20237.doi:10.1039/c3ra42634c
(31)Sousa-Aguiar,E.F.;Camorim,V.L.D.;Zotin,F.M.Z.;Correa dos Santos,R.L.Microporous Mesoporous Mat.1998,25(1),25.
(32)Sousa-Aguiar,E.F.;Trigueiro,F.E.;Zotin,F.M.Z.Catal. Today 2013,218,115.
(33)Huang,J.;Jiang,Y.J.;Reddy Marthala,V.R.;Ooi,Y.S.;Weitkamp,J.;Hunger,M.Microporous Mesoporous Mat.2007,104,129.doi:10.1016/j.micromeso.2007.01.016
(34)Li,S.H.;Zheng,A.M.;Su,Y.C.;Zhang,H.L.;Chen,L.;Yang,J.;Ye,C.H.;Deng,F.J.Am.Chem.Soc.2007,129,11161.doi:10.1021/ja072767y
(35)Lorprayoon,V.;Condrate,R.A.Appl.Spectrosc.1982,36(6),696.doi:10.1366/0003702824639259
(36)Tian,F.P.;Wu,W.C.;Jiang,Z.X.;Yang,Y.X.;Cai,T.C.;Li,C.Chem.J.Chin.Univ.2005,26(12),2351.[田福平,吴维成,蒋宗轩,杨永兴,蔡天锡,李灿.高等学校化学学报,2005,26(12),2351.]
(37)Qin,Y.C.;Gao,X.H.;Duan,L.H.;Fan,Y.C.;Yu,W.G.;Zhang,H.T.;Song,L.J.Acta Phys.-Chim.Sin.2014,30(3),544.[秦玉才,高雄厚,段林海,范跃超,于文广,张海涛,宋丽娟.物理化学学报,2014,30(3),544.]doi:10.3866/PKU. WHXB201401021
Modulation of the Acidity and Catalytic Conversion Properties of Y Zeolites Modified by Cerium Cations
ZHANG Chang1QIN Yu-Cai1GAO Xiong-Hou2,*ZHANG Hai-Tao2MO Zhou-Sheng3CHU Chun-Yu1ZHANG Xiao-Tong1SONG Li-Juan1,3,*
(1Key Laboratory of Petrochemical Catalytic Science and Technology of Liaoning Province,Liaoning ShiHua University,Fushun 113001,Liaoning Province,P.R.China;2Lanzhou Petrochemical Research Center,Petrochemical Research Institute,Petro China Company Limited,Lanzhou 730060,P.R.China;3College of Chemistry and Chemical Engineering,China University of Petroleum(East China),Qingdao 266555,Shandong Province,P.R.China)
Y-type zeolites with different cerium ion content were prepared by liquid phase ion exchange(LPIE)and their structural properties were characterized by inductively coupled plasma atomic emission spectrometry (ICP-AES),X-ray diffraction(XRD),N2adsorption isotherm,and temperature-programmed desorption of ammonia(NH3-TPD).The influence of cerium ion modification of the Y-type zeolites on the acidity and catalytic behavior was studied by in situ Fourier transform infrared spectroscopy(in situ FTIR)techniques with pyridine and thiophene as probe molecules.The results indicate that the original crystal structures of the zeolites remain unchanged after modification with cerium ions.During the modification process the Ce species tend to be located in sodalite(SOD)cages after calcination and remain in the supercages upon a gradual increase in Ce cation content.The amount and strength of the Brönsted(B)acid sites in the zeolites generated by themodification increases initially and then stabilizes with an increase in Ce ion content.The strong and weak Lewis (L)acid sites related to the non-framework aluminum and the rare earth species increase continuously during the modification process.Thiophene adsorption FTIR spectra indicate that the adsorbed thiophene molecules protonate at the strong Brönsted acid sites of the zeolites.The protonated products then oligomerize.The synergy between Ce species and B acid sites is favorable for the thiophene oligomerization reaction.
October 8,2014;Revised:December 15,2014;Published on Web:December 16,2014.
In situ Fourier transform Infrared spectroscopy;CeY;Acidity;Thiophene;Catalytic conversion
O643
10.3866/PKU.WHXB201412163
GAO Xiong-Hou,Email:gaoxionghou@petrochina.com.cn.
The project was supported by the National Natural Science Foundation of China(21076100,21376114)and Major Program of Petroleum Refining of Catalyst of Petro China Company Limited,China(10-01A-01-01-01).
国家自然科学基金(21076100,21376114)和中国石油天然气股份有限公司炼油催化剂重大专项(10-01A-01-01-01)资助项目
©Editorial office ofActa Physico-Chimica Sinica
