甘草药材HPLC 指纹图谱研究*
2015-08-10谢佳颖杨立国夏伟军张海元张英杰梅双喜
谢佳颖,杨立国,夏伟军,张海元,张英杰,梅双喜
(云南省药物研究所/云南白药集团创新研发中心/云南省中药和民族药新药创制企业重点实验室,云南 昆明650111)
甘草为豆科植物甘草Glycyrrhiza uralensis Fisch.的干燥根及根茎,春、秋二季采挖,除去须根,晒干。其性平、味甘,归心、肺、脾、胃经,具有补脾益气、清热解毒之功效,临床上广泛应用于脾胃虚弱,倦怠乏力等证[1-2]。
甘草是云南省药物研究所研制的“金品”痛舒胶囊、肿痛气雾剂等产品的重要原料之一[3],但由于药材受气候等生态环境的影响,不同批次药材所含化学成分差别较大。为了有效的控制各批次甘草药材的质量,我们运用HPLC 方法对同一产地10 批次甘草药材进行了指纹图谱测定,为甘草药材质量的有效控制奠定了方法基础。
1 仪器和材料
Thermo UltiMate 3000 高效液相色谱仪(Ultimate-3000 RS 四元泵、WPS-3000 自动进样器、TCC -3000 SD 柱 温 箱、DAD -3000 检 测 器、ChromeLeon 7.0 色谱工作站);SK8200HP 超声波清洗器(上海科导超声仪器有限公司);瑞士Mettler Toledo AG285 电子天平;甘草苷(111610-200503)购自中国食品药品检定研究院;甘草酸(130109)和异甘草苷(130418)购自北京万佳首化生物科技有限公司;芹糖甘草苷和异甘草素葡萄糖芹菜苷为本研究室从甘草药材中提取分离得到[4-5];甘草药材(Glycyrrhiza uralensis Fisch.)购自内蒙古,经云南省药物研究所邱斌高级工程师鉴定,标本保存于云南省药物研究所标本室;乙腈为色谱级,水为实验室自制重蒸水,其它试剂均为分析纯。
2 方法和结果
2.1 液相色谱条件
色谱柱为ZORBAX Eclipse XDB-C18(4.6 mm× 250 mm,5 μm),流动相为乙腈(A)-0.05%磷酸水溶液(B),梯度洗脱程序见表1。流速为1mL/min,各时间段检测波长为:0~16 min,215 nm;16~22 min,360 nm;22~70 min,250 nm,柱温为30 °C,进样量为10 μL,检测时间为70 min。

表1 流动相梯度洗脱表
2.2 混合对照品溶液的制备方法
分别称取异甘草素葡萄糖芹菜苷、异甘草苷、芹糖甘草苷、甘草苷和甘草酸适量,加入70%乙醇制成每毫升含上述各对照品20 μg 的混标溶液,摇匀,备用。
2.3 供试品溶液的制备方法
取甘草药材粉末约0.1 g,置60 mL 具塞锥形瓶中,加入70%乙醇25 mL,密塞,称重,超声处理(功率250 W,频率40 kHz)30 min,放冷,再称定重量,加入70%乙醇补足减失的重量,摇匀后,使用0.45 μm 微孔滤膜滤过,取续滤液,备用。
2.4 方法学验证
2.4.1 精密度考察
取1 号供试品溶液,连续进样6 次,记录高效液相色谱图,计算各个共有峰的相对保留时间和相对峰面积,计算RSD 值。各共有峰相对保留时间的RSD 为0.02%~0.35%(表2),各共有峰相对峰面积的RSD 为0.03%~0.13%(表3),表明仪器性能较好,符合指纹图谱相关要求。

表2 相对保留时间(将3 号峰的相对保留时间定为1.000)

表3 相对峰面积(将3 号峰的相对峰面积定为1.000)
2.4.2 稳定性试验
取1 号供试品溶液,分别在制备后的0,2,4,8,12,24 h 进样,记录高效液相色谱图,计算各共有峰的相对保留时间和相对峰面积,计算RSD 值。各共有峰相对保留时间的RSD 为0.05%~1.84%(表4),各共有峰的相对峰面积的RSD 为0.02%~0.83%(表5),表明供试品溶液在24 h 内稳定,符合指纹图谱相关要求。
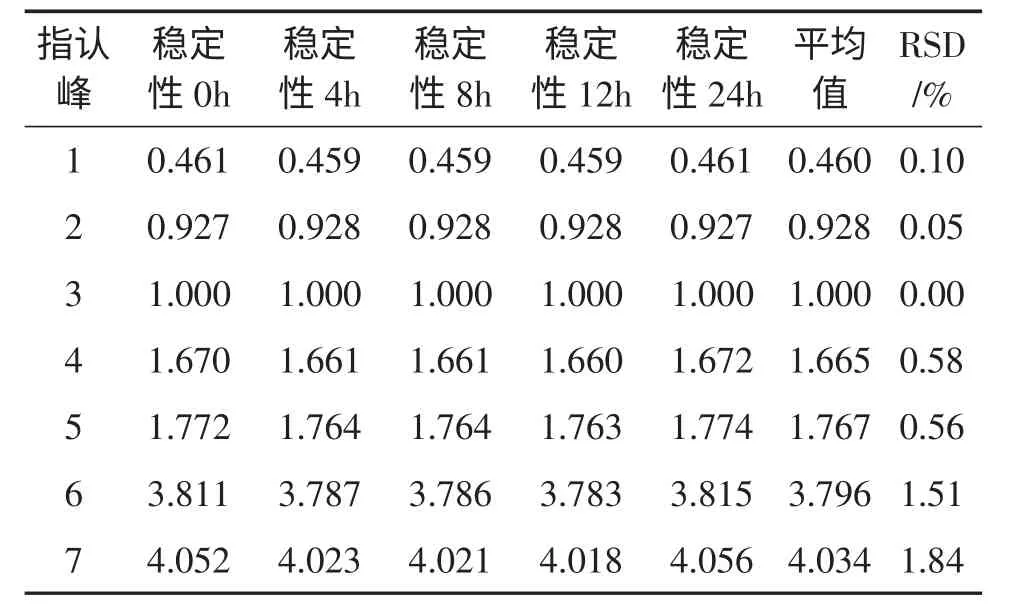
表4 相对保留时间(将3 号峰的相对保留时间定为1.000)

表5 相对峰面积(将3 号峰的相对峰面积定为1.000)
2.4.3 重复性试验
取1 号甘草药材,按2.3 项下供试品的制备方法制备6 份供试品溶液,分别进样,计算各共有峰的相对保留时间和相对峰面积,计算RSD 值。各共有峰相对保留时间的RSD 值为0.05%~1.47%(表6),各共有峰相对峰面积的RSD 值为0.05%~0.36%(表7),表明供试品溶液的制备方法重复性较好,符合指纹图谱相关要求。

表6 相对保留时间(将3 号峰的相对保留时间定为1.000)

表7 相对峰面积(将3 号峰的相对峰面积定为1.000)
2.5 指纹图谱及相似度计算
2.5.1 十批甘草药材指纹图谱的测定
按2.1 项下HPLC 条件测定同一产地10 批次甘草药材的高效液相色谱指纹图谱,采用药典委员会推荐使用的“中药色谱指纹图谱相似度评价系统”软件进行分析,得到了不同批次甘草药材指纹图谱的共有谱图,并确定了7 个共有色谱峰(见图1)。
2.5.2 同一产地10 批次甘草药材指纹图谱相似度计算
采用药典委员会推荐使用的“中药色谱指纹图谱相似度评价系统”软件,计算得到了同一产地10批次甘草药材指纹图谱的相似度,其相似度均在0.95 以上,表明同一产地不同批次甘草药材的相似度满足要求(见图2)。
2.6 甘草药材部分特征峰的化学指认
取1 号供试品溶液及混合对照品溶液进样,根据各个对照品的色谱保留时间可以对甘草药材的部分色谱峰进行指认,峰2 为芹糖甘草苷,峰3 为甘草苷,峰4 为异甘草素葡萄糖芹菜苷,峰5 为异甘草苷,峰6 为甘草酸(见图3)。

图1 甘草药材HPLC 对照指纹图谱

图2 同一产地10 批次甘草药材HPLC 指纹图谱

图3 混合对照品(上图)和甘草药材(下图)的HPLC 图谱
3 讨论
①本研究对甲醇、乙腈和酸水系统进行了摸索,发现乙腈-0.05%磷酸水系统分离情况较好,各色谱峰均达到比较好分离,故确定乙腈-0.05%磷酸水系统作为甘草药材液相色谱指纹图谱的流动相。
②实验过程中考察了3 种不同提取方式(超声、冷浸和回流),发现3 种提取方式的提取效率相似,故最终选择操作简便的超声作为药材的提取方式。
③根据二极管阵列检测器全波长扫描给出的各指标成分的最大吸收波长可知:芹糖甘草苷和甘草苷(2 号峰和3 号峰)的最大吸收波长为215 nm,异甘草素葡萄糖芹菜苷和异甘草苷(4 号峰和5 号峰)的最大吸收波长为360 nm,甘草酸(6 号峰)的最大吸收波长为250 nm。所以,确定各时间段的检测波长如下:0~16 min,215 nm;16~22 min,360 nm;22~70 min,250 nm。
④甘草药材中的主要化学成分包括黄酮类、三萜类、多糖类和生物碱类[6-10],具有镇咳平喘、抗肿瘤、抗炎、抗病毒等作用[11-15]。上述黄酮类和三萜类化学成分很可能是云南省药物研究所研制的“金品”痛舒胶囊、肿痛气雾剂等产品的活性成分,本研究所建立的甘草药材的HPLC 指纹图谱方法,对上述黄酮类和三萜类化学成分进行了测定,可用于甘草药材的质量控制,为实现“金品”痛舒胶囊、肿痛气雾剂等产品质量的均一、稳定奠定了方法基础。
[1] 国家药典委员会. 中华人民共和国药典2010 年版(一部)[M]. 北京:中国医药科技出版社,2010:80-81.
[2] 张翠英,常段玲,周应群,等. 乌拉尔甘草水溶性成分的HPLC 指纹图谱研究[J]. 北京中医药大学学报,2009,32(12):842-845.
[3] 云南省药物研究所. 云南省药物研究所制药厂金品系列药品临床推介[J]. 云南中医中药杂志,2004,25(5):50.
[4] Kitagawa I,Chen WZ,Hori K,et al. Chemical studies of Chinese Licorice-Roots. Evaluation of five new flavonoid constituents from the roots of Glycyrrhiza glabra L. collected in Xinjiang [J]. Chem. Pharm. Bull,1994,42(5):1056-1062.
[5] 白虹,窦德强,裴玉萍,等. 栽培甘草的化学成分研究[J].中草药,2005,36(5):652-654.
[6] 张永,严安定,高建. 液质联用技术鉴定甘草提取物中的主要化学成分[J]. 中成药,2012,34(6):1111-1115.
[7] 惠寿年,董阿玲. 国内对甘草化学成分的研究进展[J]. 中草药,1999,30(4):313-315.
[8] 梁冰,杨爱馥,黄凤兰,等. 甘草属化学成分及药理作用研究进展[J]. 东北农业大学学报,2006,37(1):115-119.
[9] 彭励,胡正海. 甘草生物学及化学成分的研究进展[J]. 中草药,2005,36(11):1744-1747.
[10] 李薇,宋新波,张丽娟,等. 甘草中化学成分研究进展[J].辽宁中医药大学学报,2012,14(7):40-44.
[11] 贾国惠,贾世山. 甘草中黄酮的药理作用研究进展[J]. 中国药学杂志,1998,33(9):513-516.
[12] 王访,苏耀海. 甘草的药理作用及临床应用[J]. 时珍国医国药,2002,13(5):303-304.
[13] 李德芳,王振华,罗锋,等. 异甘草素的药理作用研究[J].时珍国医国药,2010,21(2):362-364.
[14] 张玉龙,王梦月,杨静玉,等. 炙甘草化学成分及药理作用研究进展[J]. 上海中医药大学学报,2015,29(3):99-102.
[15] 陈超,李宁,倪慧,等. 甘草化学成分分离、细胞培养和分析研究进展[J]. 现代药物与临床,2011,26(3):188-194.
