纳米SiO2表面接枝聚环氧丙醇及其对聚氨酯泡沫力学性能的影响
2015-07-18陈可平王建华梁书恩黄奕刚田春蓉
高 霞 陈可平 王建华 梁书恩 黄奕刚 田春蓉
(1.西南科技大学材料科学与工程学院 四川绵阳 621010;2.中国工程物理研究院化工材料研究所 四川绵阳 621900)
聚氨酯硬质泡沫塑料是一种集质轻、抗振、噪声低等多种功能于一体的泡沫材料[1],主要用作保温隔热、抗冲击及缓冲吸能等。通过调整配方[2-3],改变原料规格,制备不同密度、硬度且具有阻燃、电学等特殊性能的硬泡制品,可满足不同需求和应用条件。近年来,随着应用领域的不断拓宽和环保意识的增强,聚氨酯泡沫(polyurethane foam,PUF)材料显示了其在力学性能、耐热、抗菌以及电学等方面的不足[1,4]。为弥补其性能缺陷和应对发展的新趋势,最有效的方法之一是在聚氨酯体系中引入无机纳米粒子[5]。纳米 SiO2(nano silica,NS)具有多孔性、质轻、耐高温、不燃烧等优异性能,并具有良好的化学稳定性和电绝缘性,在PUF中引入纳米SiO2,有良好的力学增强作用[6]。然而,单纯引入无机纳米SiO2具有以下缺点:纳米SiO2与有机PUF之间界面不相容、密度差异大,造成难以均匀稳定地分散而导致增强效果不佳且泡沫材料性能不均匀。研究表明:对无机纳米粒子表面修饰[7],可改善其与聚合物基体间的相容性,进而实现其在聚合物基体中均匀稳定地分散。其中,表面接枝聚合物具有更好的分子可设计性和结构可控性,因此可根据不同聚合物基体选择接枝与其相容性较好的聚合物,制备出具有各种独特性能的新材料,满足不同领域的特殊要求[7]。
引发接枝反应的方法较多,选择性灵活,包括原子转移自由基聚合(ATRP)[8]、可逆加成/断裂链转移聚合(RAFT)[9]、阴阳离子聚合[10]、自由基聚合[11-12]、过 渡 金 属 催 化 聚 合 以 及 开 环 歧 化 聚合[13-14]等。Wilhelm T.S.Huck 等通过阴离子开环聚合法,利用甲醇钠先质子化纳米 SiO2表面Si-OH,然后在110℃条件下,作为引发剂攻击环氧丙醇单体的未取代侧,形成二次醇盐,继续攻击其它单体或与相邻的伯醇交换质子,在SiO2表面接枝聚环氧丙醇[10]。
本文采用阴离子聚合法在纳米SiO2表面原位接枝聚环氧丙醇,制备了纳米SiO2复合PUF,并研究了纳米SiO2接枝前后对复合PUF的泡孔结构和压缩性能的影响。
1 实验
1.1 试剂
正硅酸四乙酯,分析纯,国药集团化学试剂有限公司;无水乙醇、氨水、甲苯和无水甲醇,均为分析纯,成都市联合化工试剂研究所;金属钠,分析纯,天津市科密欧化学试剂有限公司;环氧丙醇,分析纯,阿拉丁试剂有限公司;聚醚多元醇:N303(OH值458.2 mg/g),工业级,江苏金栖聚氨酯有限公司;聚芳基聚亚甲基异氰酸酯(poly aryl polymethylene isocyanate,PAPI):NCO content=31.2%(质量分数),工业级,烟台万华聚氨酯有限公司;催化剂:三乙醇胺,分析纯,重庆川东化工(集团)有限公司;发泡剂:蒸馏水;泡沫稳定剂:硅油AK8807,工业级,南京德美世创化工有限公司。
1.2 球形纳米SiO2的制备
将无水乙醇(900 mL),蒸馏水(72.00 g),氨水(25.97 g)依次加入反应瓶内,在室温下500 r/min搅拌10 min,加入正硅酸四乙酯124.88 g和无水乙醇100 mL,继续搅拌3 h,高速离心去除多余溶剂,用蒸馏水洗涤5次,经真空干燥得到纳米SiO2。
1.3 纳米SiO2表面原位接枝聚环氧丙醇
取100 mL无水甲醇倒入三口烧瓶,氮气保护,室温下以400 r/min搅拌,加入金属钠待其反应完全,得到0.34 mol/L甲醇钠溶液。然后,称取上述纳米SiO25.06 g至三口烧瓶内,超声震荡10 min,继续反应1 h。停止反应后,采用无水甲醇(20 mL×2)、经金属钠蒸馏得到的甲苯(20 mL×2)洗涤,再干燥、研磨得到表面醇钠处理的纳米SiO2。
称取上述处理后纳米SiO24.90 g,环氧丙醇经分子筛除水(10.12 g),加入三口烧瓶,搅拌速度400 r/min,温度110℃,氮气保护,反应45 min后离心分离,并用乙醇和水的混合溶液(30 mL×4)洗涤,经干燥得到表面接枝聚环氧丙醇的纳米SiO2[10]。
1.4 制备纳米SiO2复合PUF
按配方设计称取蒸馏水、三乙醇胺、硅油AK8807加入定量聚醚N303中,然后加入不同填充量接枝前后的纳米SiO2(与聚醚质量比为0%,0.5%,1%,1.5%,2%),用机械搅拌器快速搅拌均匀,控制混合物温度25℃,加入一定量的PAPI,继续搅拌2 min,至杯底微微发热,混合物开始发白后倒入模温为40℃的模具中,待泡沫反应完全后,放入100℃的烘箱中,固化2~3 h,脱模,制得纳米SiO2复合PUF。
1.5 测试与表征
红外(FTIR)分析:使用Thermo Fisher公司的Nicolet 6700型红外光谱仪,采用溴化钾压片法测试。
X射线光电子能谱(XPS)分析:使用美国Thermo VG Scientific公司的VG-ESCALAB-250型X射线光电子能谱仪测试。
热重分析:采用美国TA公司的TA2050热重分析仪,样品在N2气氛中,100℃恒温5 min除去水分,再50℃以20℃/min升温至800℃,进行测试。
纳米SiO2表面形貌及泡沫微观形貌与结构分析:采用英国CAMSCAN公司的Apollo 300型扫描电子显微镜观察,样品以低浓度分散在无水乙醇中并滴加在硅片上;PUF样品取自然断裂面为观察面,均经喷金制得,泡孔尺寸通过Image-Pro Plus软件分析。
压缩性能分析:依据 GB/T 8813-2008,采用Instron 5500电子万能实验机进行压缩性能测试,试样尺寸为Φ 20 mm×20 mm,横梁速度为5 mm/min。
2 结果与讨论
2.1 纳米SiO2接枝原理及接枝前后红外光谱分析
在纳米SiO2表面采用阴离子开环聚合法接枝聚环氧丙醇的反应原理如图1所示。纳米SiO2表面Si-OH被甲醇钠溶液质子化,作为引发剂攻击环氧丙醇单体的未取代侧,形成二次醇盐,继续攻击其它单体或与相邻的伯醇交换质子,从而实现环氧丙醇在纳米SiO2表面的开环聚合。
纳米SiO2接枝前后红外谱图如图2所示,接枝前(曲线b)在1100 cm-1有明显O-Si-O特征吸收,并在3300 cm-1出现-OH特征吸收,说明已成功合成 SiO2。接枝后(曲线 a)在 1450 cm-1,2930 cm-1处产生新的谱峰,分析为对应聚环氧丙醇中 C -O -C,-CH - 的伸缩振动[10],同时原3300 cm-1处-OH特征吸收也明显增强,是由于接枝产物具有更多的-OH造成。因为样品在水和乙醇中超声振荡并经高速离心分离、反复洗涤,所以不是其表面吸附的环氧丙醇单体及自聚物的影响,因此说明在SiO2表面已成功接枝聚环氧丙醇。
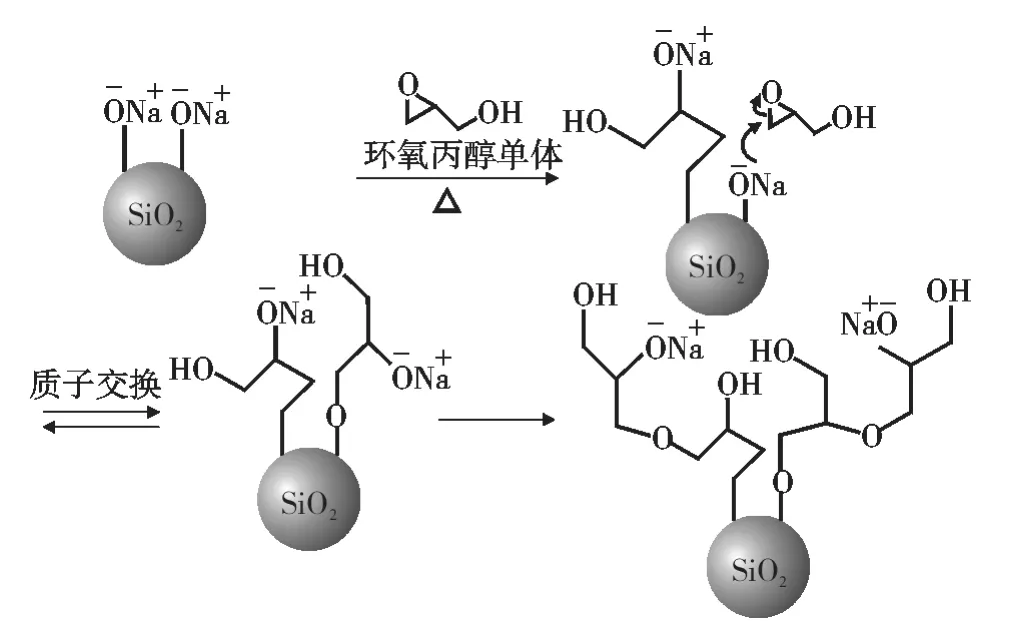
图1 纳米SiO2表面接枝聚环氧丙醇反应原理示意图Fig.1 The reaction pathways of the grafting polyglycidol on the surface of nano SiO2(NS)
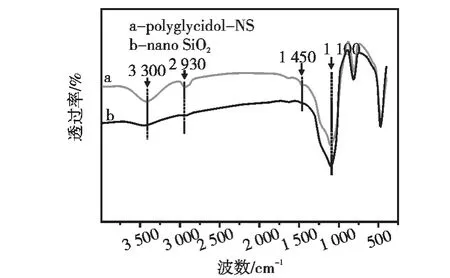
图2 纳米SiO2红外光谱图Fig.2 Infrared spectra of nano SiO2(NS)samples
2.2 纳米SiO2接枝前后X射线光电子能谱分析
纳米SiO2接枝前后X射线光电子能谱图如图3所示。图3(a)为全谱图,在全谱图中,接枝前,Si2p(103.41 eV),O1s(530.81 eV)的信号峰清晰可见,均归属为 SiO2的结合能,说明 SiO2的存在;C1s(282.66 eV)信号峰,推测是由测试过程中不可避免的含碳杂质造成。接枝后,新增Na1s信号峰,并且C1s,O1s信号峰也发生偏移。图3(b)为 C1s高分辨扫描图,由图3(b)可见,接枝后,C1s信号峰偏移至286.61 eV,可归属为C-O-C,C-OH,-CH2-结合能,与聚环氧丙醇所含基团相吻合。图3(c)为Na1s高分辨扫描图,与接枝前相比,接枝后纳米SiO2新增Na1s(1071.69 eV)信号峰,说明含有用于质子化的Na+。图3(d)为O1s高分辨扫描图,接枝后纳米SiO2谱图中O1s信号峰偏移至532.77 eV,归属为C-O-C结合能,可推测为接枝产物中含有的C-O-C。因此,进一步说明已成功接枝聚环氧丙醇[15]。
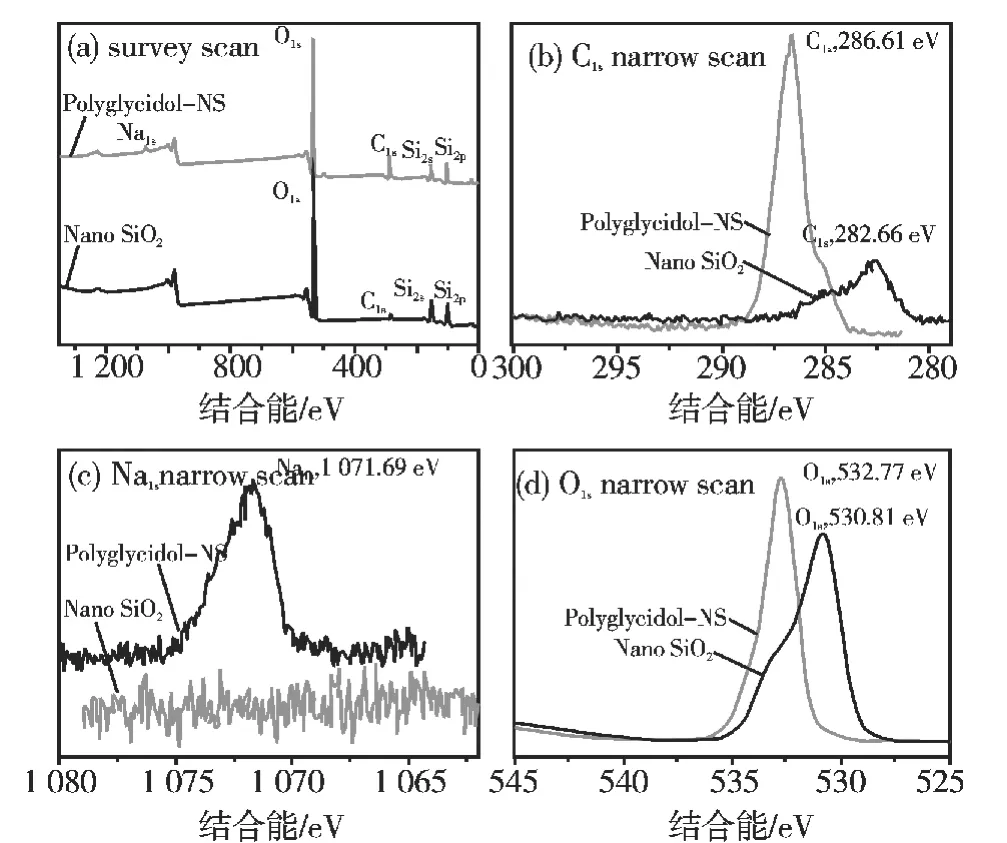
图3 纳米SiO2X射线光电子能谱图Fig.3 X -ray photoelectron spectra of nanoSiO2(NS)samples
2.3 热重分析
纳米SiO2接枝前后热重分析结果如图4所示,接枝前(曲线a),纳米SiO2在100~600℃产生约4%(质量分数,下同)失重,分析认为是纳米SiO2表面吸附的水分及合成过程导致的溶剂残留造成的;接枝后(曲线b)在300~550℃有明显失重,相比接枝前约6%,表明在该温度范围内其表面的聚环氧丙醇发生了热分解,接枝量较低是因为环氧丙醇单体直接在纳米SiO2表面聚合的同时会发生自聚,形成低聚物,且能以该形式接枝在纳米SiO2表面,由于空间位阻效应,限制了更多单体在其表面接枝,从而限制了聚环氧丙醇的接枝率[16]。
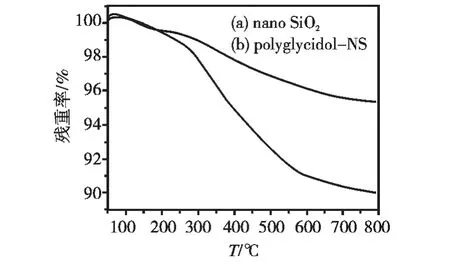
图4 纳米SiO2热失重曲线Fig.4 TG analysis of nano SiO2samples
2.4 扫描电镜(SEM)分析
图5是纳米SiO2接枝前后SEM图。图5(a)显示,合成的SiO2呈球形,粒径分布较均匀,有团聚现象出现。由图5(b)可见,合成的SiO2粒径为100~150 nm,粒径稍有波动是因为反应过程中各部位所受机械搅拌作用不同所致。图5(c)对比图5(a)可见,纳米SiO2接枝后仍呈球形,粒径无明显变化,可均匀分散。由图5(d)可见,接枝后纳米SiO2表面明显有一层毛绒状物,推测其为所接枝聚合物,从形貌上说明已成功接枝聚环氧丙醇。

图5 纳米SiO2的扫描电镜图Fig.5 SEM images of nano SiO2samples
2.5 纳米SiO2接枝前后对PUF泡孔结构的影响

图6 纳米SiO2接枝前后不同填充量复合PUF扫描电镜图Fig.6 SEM images of PUF with different nano SiO2content
图6为纳米SiO2接枝前后不同填充量复合PUF泡孔结构的SEM照片。图6(b)与图6(a)相比,填充纳米SiO2后PUF孔径减小,泡孔密度增大,从而加强机械作用,这是因为纳米SiO2增加了反应过程中的成核位点[16]。图6(d)显示,填加量为2 php时,泡孔孔径反而增大,且分布极不均匀。这是由于纳米SiO2达一定量后,成核位点过多[16],泡孔之间相互拥挤发生并泡,造成泡孔或大或小,而且,由于纳米SiO2过多,团聚严重,甚至破坏泡孔结构,出现碎泡、裂泡[17]。纵观图 6(a)、图 6(b)、图 6(d),随着纳米SiO2填加量增加,泡孔孔径先减小后增大,泡孔结构也随之先优化后劣化。该规律同样适用接枝后纳米SiO2复合PUF,但是,相同填加量时,接枝后纳米SiO2复合PUF泡孔孔径更小,分布更均匀,泡孔结构更饱满、圆滑,且填加量加至一定量时,仍能保持较均匀的孔径分布,且显著减少碎泡、裂泡(图6e)。这是因为表面接枝的聚环氧丙醇具有阻隔作用,能有效阻止纳米SiO2在与聚醚混合过程中相互团聚,同时由于与聚醚具有相同基团,可提高其与聚醚相容性[18]。
2.6 纳米SiO2接枝前后对聚氨酯泡沫压缩性能的影响
表1为纳米SiO2不同填充量复合PUF的压缩性能数据(比强度:压缩强度/试样密度)。由表1可知,随着初始纳米SiO2填充量增加,复合PUF压缩模量和比强度随之增加,原因是纳米SiO2与PUF基体间相互作用,约束了聚氨酯链段的运动[18-19],且有效地促进了粒子与基体间的荷载转移[16],同时纳米SiO2作为成核剂,减小泡孔尺寸[20]。而SiO2填充量过多,容易团聚[21-22],与 PUF 基体间相互作用减弱,泡孔尺寸增大,甚至出现碎泡,所以填充量为2 php时,复合PUF压缩性能略有下降。

表1 纳米SiO2接枝前后不同填充量复合PUF的压缩性能Table 1 The compressive properties of PUF with different nano SiO2contents
接枝后纳米 SiO2改性的PUF,填充量0.5 php时,压缩模量从243 MPa提高到295 MPa,比强度从17.52(MPa·cm3·g-1)提高到19.33(MPa·cm3·g-1)。高填充量(1,1.5,2 php)时,压缩性能反而下降。可能的原因是接枝后纳米SiO2表面的聚环氧丙醇起到阻隔作用,减少粒子间的团聚,且与聚醚具有相同的基团,从而与PUF基体相容性更好,相互作用也更紧密。填充量过高时,可能也会因粒子接有相同聚合物而吸引作用增强,导致团聚,降低复合PUF压缩性能。
3 结论
(1)本文采用阴离子开环聚合法,在纳米SiO2表面原位接枝聚环氧丙醇,通过红外光谱及X射线光电子能谱分析说明接枝成功;扫描电镜结果显示,接枝后纳米SiO2仍呈规则球形,表面有毛绒状物,粒径无明显变化,约为120 nm;热重分析证明接枝量为6%。
(2)接枝前纳米SiO2复合PUF泡孔孔径随着填充量的增加而减小,当填充量高于1.5 php时,反而增大;相比接枝前,接枝后纳米SiO2复合PUF泡孔结构得到优化,孔径也相对减小,填充量为2 php时,平均孔径从 276.90 μm 减小到 197.45 μm。
(3)接枝前纳米SiO2复合PUF力学性能随着填充量的增加而改善。与接枝前相比,接枝后纳米SiO2复合PUF力学性能在低填充量更优异,填充量为0.5 php 时,比强度提高约1.81(MPa·cm3·g-1),压缩模量提高约52 MPa。
[1]SILVA M C,TAKAHASHI J A,CHAUSSY D,et al.Composites of rigid polyurethane foam and cellulose fiber residue[J].Journal of Applied Polymer Science,2010,177(6):3665-3672.
[2]YOU K M,PARK S S,LEE C S,et al.Preparation and characterization of conductive carbon nanotube-polyurethane foam composites[J].Journal of Materials Science,2011,46(21):6850-6855.
[3]XIA H,SONG M,JIN J,et al.Poly(propylene glycol)-Grafted Multi-Walled Carbon Nanotube Polyurethane[J].Macromolecular Chemistry and Physics,2006,207(21):1945-1952.
[4]XU Z,TANG X,GU A,et al.Novel preparation and mechanical properties of rigid polyurethane foam/organoclay nanocomposites[J].Journal of Applied Polymer Science,2007,106(1):439-447.
[5]ALAVI NIKJE M M,FARAHMAND NEJAD M A,SHABANI K,et al.Preparation of magnetic polyurethane rigid foam nanocomposites[J].Colloid and Polymer Science,2012,291(4):903 -909.
[6]YANG Z G,ZHAO B,QIN S L,et al.Study on the mechanical properties of hybrid reinforced rigid polyurethane composite foam[J].Journal of Applied Polymer Science,2004,92(3):1493 -1500.
[7]LIU J,LIU F,GAO K,et al.Recent developments in the chemical synthesis of inorganic porous capsules[J].Journal ofMaterialsChemistry, 2009, 19(34):6073-6084.
[8]LIU T,JIA S,KOWALEWSKI T,et al.Grafting poly(n-butyl acrylate)from a functionalized carbon black surface by atom transfer radical polymerization[J].Langmuir,2003,19(16):6342 -6345.
[9]ROWE M D,HAMMER B A,BOYES S G.Synthesis of surface-initiated stimuli-responsive diblock copolymer brushes utilizing a combination of ATRP and RAFT polymerization techniques[J]. Macromolecules, 2008,41(12):4147-4157.
[10]KHAN M,HUCK W T.Hyperbranched polyglycidol on Si/SiO2surfaces via surface-initiated polymerization[J].Macromolecules,2003,36(14):5088-5093.
[11]RANJAN R,BRITTAIN W J.Combination of living radical polymerization and click chemistry for surface modification[J]. Macromolecules, 2007, 40(17):6217-6223.
[12]BARBEY R,LAVANANT L,PARIPOVIC D,et al.Polymer brushes via surface-initiated controlled radical polymerization:synthesis,characterization,properties,and applications[J].Chemical Reviews,2009,109(11):5437-5527.
[13]JOUBERT M,DELAITE C,BOURGEAT‐LAMI E,et al.Hairy PEO‐Silica Nanoparticles through Surface‐Initiated Polymerization of Ethylene Oxide[J].Macromolecularrapidcommunications, 2005, 26(8):602-607.
[14]JOUBERT M,DELAITE C,BOURGEAT‐LAMI E,et al.Ring‐ opening polymerization of ε ‐ caprolactone and L ‐ lactide from silica nanoparticles surface[J].Journal of Polymer Science Part A:Polymer Chemistry,2004,42(8):1976-1984.
[15]XIA H,SONG M,JIN J,et al.Poly(propylene glycol)-grafted multi-walled carbon nanotube polyurethane[J].Macromolecular Chemistry and Physics,2006,207(21):1945-1952.
[16]BOURGEAT-LAMI E,LANG J.Effect of silica size and concentration on the morphology of silica-polystyrene composite particles[J].Journal of Colloid and Interface Science,1999,210(2):281-289.
[17]KANG J W,KIM J M,KIM M S,et al.Effects of nucleating agents on the morphological,mechanical and thermal insulating properties of rigid polyurethane poams[J]. MacromolecularResearch,2009,17(11):856-862.
[18]LIANG J,HUANG Y,ZHANG L,et al.Molecular ‐level dispersion of graphene into poly(vinyl alcohol)and effective reinforcement of their nanocomposites[J].AdvancedFunctionalMaterials, 2009, 19(14):2297-2302.
[19]LIU T,PHANG I Y,SHEN L,et al.Morphology and mechanical properties of multiwalled carbon nanotubes reinforced nylon-6 composites[J].Macromolecules,2004,37(19):7214-7222.
[20]WANG H,BAI Y,LIU S,et al.Combined effects of silica filler and its interface in epoxy resin[J].Acta Materialia,2002,50(17):4369 -4377.
[21]KARTHIKEYAN C,SANKARAN S,KUMAR J.Processing and compressive strengths of syntactic foams with and without fibrous reinforcements[J].Journal of applied polymer science,2001,81(2):405 -411.
[22]RAFIEE M A,RAFIEE J,WANG Z,et al.Enhanced mechanical properties of nanocomposites at low graphene content[J].ACS nano,2009,3(12):3884 -3890.
