Labdane二萜“类天然产物”的合成研究
2015-07-18张军庆刘跃佳宁弘历杨海君
张军庆 刘跃佳 宁弘历 廖 盼 杨海君
(西南科技大学材料科学与工程学院 四川绵阳 621010)
天然产物骨架的复杂性和丰富的官能团化赋予了天然产物类化合物独有的生物学活性,因此天然产物作为药物研究的先导化合物有其无法替代的独特性质。随着高通量筛选技术的发展、人类基因组测序工作的完成以及近年来化学生物学研究的深入,迫切需要获得大量骨架多样、构造复杂、立体化学丰富的天然产物或者天然产物类似物,以便更快、更好地进行药物发现和化学生物学研究。以天然产物为起点,建立分子多样性和结构复杂性的“类天然产物”化合物库已成为一种必然趋势[1-5]。很多活性天然产物本身在自然界的储量有限,分离纯化困难,因而常常通过合成的方法来获取。传统的有机合成化学是以完成天然产物的定向合成为唯一目的,因此合成策路往往只适用于单个或者少数几个天然产物的全合成;化学合成存在路线长、收率低、复杂操作等问题,很难用于结构多样的天然产物或其类似物的合成。总的来说,运用传统的逆向合成分析实现的目标定向合成方法已不能满足高通量筛选和化学生物学等对化合物结构多样性的要求。为此,美国哈佛大学的 Stuart L.Schreiber教授提出了多样性导向合成的概念[6]。天然有机合成化学的研究重心已经开始从“合成单一天然产物”为目的的目标导向合成转移到“结构多样性的类天然产物”的导向性合成[7-8]。用多样性导向合成方法建立骨架多样、构造复杂、立体化学多样的“类天然产物”化合物库,将在生物学的基础研究和药物研究中起到关键的作用。
含呋喃环的labdane二萜化合物是一类从爵床科、姜科、半日花科、菊科等植物中分离出来的天然产物[9](结构式见图1),该类化合物通常都具有细胞毒活性、抗炎、抗菌等广泛的生物活性[10],但是在天然资源中的含量比较低,分离提取困难,所能获得的量有限,不能满足生物学活性等研究,因此受到广大合成化学家的广泛关注。这些化合物的结构复杂,并且具有多个手性中心,不对称合成主要通过香芹酮等来半合成[11-12]。由于半合成原料结构的限制,很难实现产物的结构多样性。全合成则主要采用多烯环化等方法来合成[13-17],但其合成路线存在路线长、产率低、立体选择性差等问题,也不易获得结构类型各异的衍生物,针对这些天然产物进行药物研发,需要获得一系列结构各异的天然产物或者“类天然产物”,对于药物发现和化学生物学研究等都极为重要[18]。
香紫苏内酯有着与含呋喃环的labdane型二萜化合物的十氢萘片段相类似的取代基和手性中心,因而可以作为半合成的原料对合成labdane型二萜化合物及其类似物进行初步的条件探索,基于此,本文拟以香紫苏内酯为原料,通过内酯开环、黄鸣龙还原等反应得到含不同芳环的labdane型二萜类似物,为后期生物活性筛选打下基础。
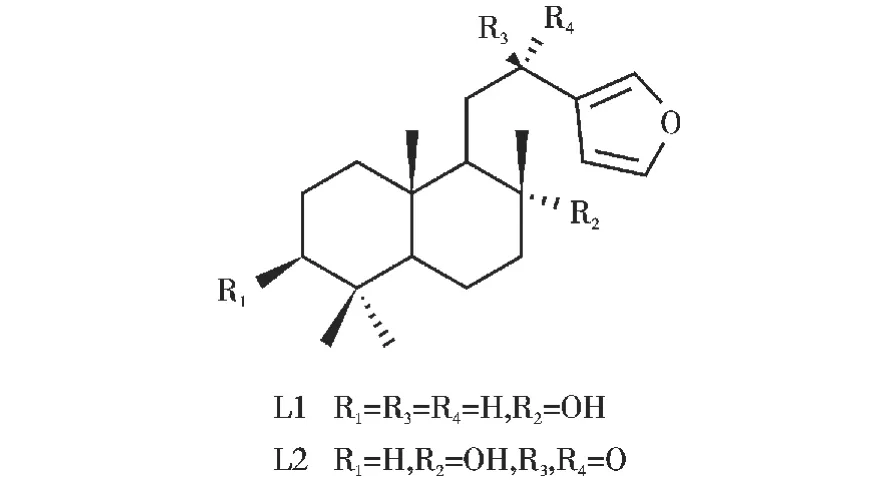
图1 部分含呋喃环的labdane二萜化合物结构式Fig.1 Some labdane diterpenes with a furan unit
1 实验部分
1.1 仪器与试剂
傅里叶变换红外光谱仪Spectrum One型(美国PE仪器公司),液相色谱-质谱联用仪Varian1200(美国瓦里安公司),600兆赫兹核磁共振谱仪Bruker Aance600(瑞士布鲁克公司),Ry-1G熔点仪(南京科航实验仪器有限公司)。
所用试剂均为市售分析纯或化学纯,四氢呋喃经除水处理,其余试剂使用前未作进一步处理。
1.2 实验过程
以香紫苏内酯为原料,通过开环内酯环反应得到1(a-c),进一步通过黄鸣龙还原反应得到2(a-c)(见图1)。
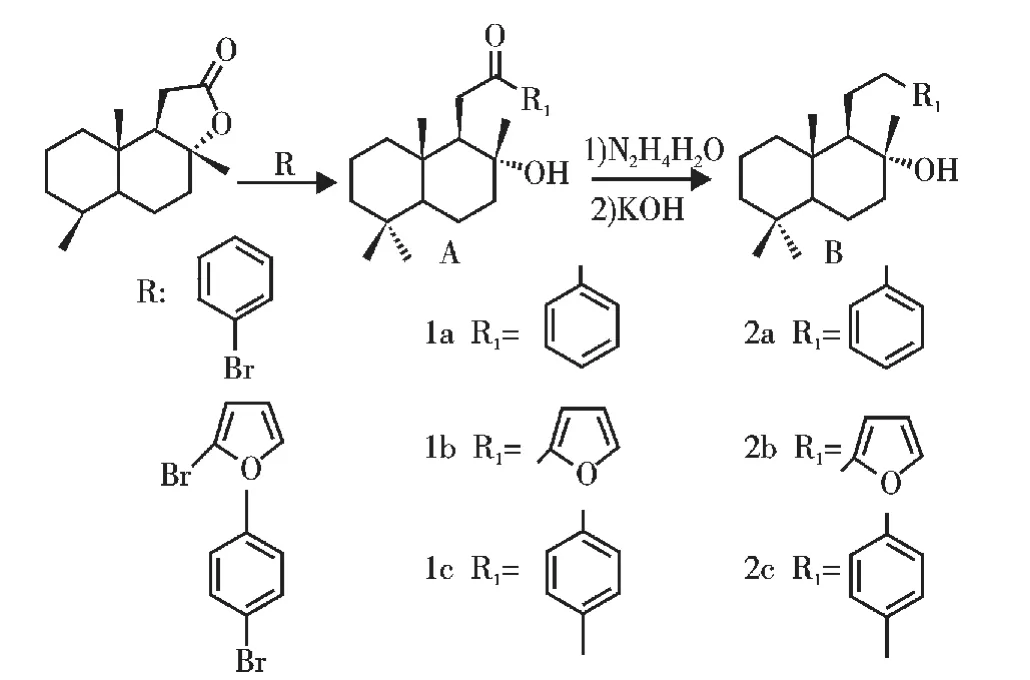
图2 以香紫苏内酯合成Labdane类天然产物Fig.2 Synthesis of labdane- like compounds starting from sclareolide
1.2.1 化合物1a的合成
在N2保护下,将0.96 g镁条加入到圆底烧瓶中,然后加入5 mL无水THF和少许碘粒,搅拌下,缓慢滴加2.1 mL溴苯的THF(10 mL)溶液,滴加完毕后,加热到40℃反应1 h,然后将所制备的格式试剂在冰浴下冷却至0~5℃,待用。将1.00 g(0.004 mol)香紫苏内酯的10 mL THF溶液,滴加到上述格式试剂中。滴加完毕后,在0~5℃下继续搅拌反应15 min,加水淬灭反应。过滤,用THF洗涤固体,乙酸乙酯萃取,合并有机相并用无水硫酸镁干燥,过滤,旋蒸,经硅胶柱层析(V石油醚/V乙酸乙酯=20/1),得到白色固体1.12 g,产率85%,mp:132~134℃;APCI-MS,m/z:328.2,[M - H]-,327.1;1H NMR(600 MHz,CDCl3):δ 7.93(d,J=6 Hz,2H),7.48(t,J=7.2 Hz,1H),7.39(t,J=7.2 Hz,2H),3.04(dd,J1=18 Hz,J2=6 Hz,1H),2.92(dd,J1=18 Hz,J2=6 Hz,1H),2.16(t,J=4.8 Hz,1H),1.89 ~1.92(td,J1=12.6 Hz,J2=3.6 Hz,1H),1.62 ~1.66(br,d,J=13.8 Hz,1H),1.23 ~ 1.50(m,6H),1.11(s,3H),1.05 ~ 1.10(m,1H),1.00 ~1.02(dd,J1=12 Hz,J2=2.4 Hz,1H),0.85 ~0.90(dt,J1=13.2 Hz,J2=3.6 Hz,1H),0.82(s,6H),0.73(s,3H);IR(KBr)v/cm-1:3386,3059,2935,2868,1673,1596,1447,1387,756,699。
1.2.2 2-溴代呋喃的制备[19]
将16.3 mL(0.225 mol)呋喃加入到 40 mL DMF中。然后在45 min内滴加20 g(0.112 mol)NBS的DMF(60 mL)溶液,滴加过程中保持温度在25~35℃。滴加完毕后,在该温度下继续搅拌反应4 h,然后蒸馏收集48~55℃之间的馏分,无水硫酸镁干燥,过滤,得到产品5.2 mL,产率57%。
1.2.3 化合物1b的合成
在-78 ℃下,N2保护下,将4.00 mL(8.7 mmol)n-BuLi(2.2 M的正己烷溶液)注射到反应瓶中,再将0.87 mL(9.2 mmol)2-溴代呋喃注射到反应瓶中,搅拌反应 10 min。然后将1.00 g(0.004 mol)香紫苏内酯的10 mL THF溶液缓慢注射到上述反应液中,在-78℃下反应45 min,自然升温至室温反应1.5 h。用10 mL饱和氯化钠溶液淬灭反应,乙酸乙酯萃取,有机相用无水硫酸镁干燥,过滤,旋蒸,所得产品经硅胶柱层析(V石油醚/V乙酸乙酯=20/1)得白色产品0.7 g,产率55%。mp:124~126℃;APCI-MS,m/z:318.2,[M - H]-,316.9;1H NMR(600 MHz,CDCl3):δ 7.51(dd,J1=1.2 Hz,J2=0.6 Hz,1H),7.16(d,J=6 Hz,1H),6.46(q,J=1.8 Hz,1H),2.90(dd,J1=17.4 Hz,J2=4.8 Hz,1H),2.82(dd,J1=17.4 Hz,J2=5.4 Hz,1H),2.06(t,J=4.8 Hz,1H),1.88 ~ 1.91(td,J1=12 Hz,J2=3.6 Hz,1H),1.62 ~ 1.66(br,d,J=13.8 Hz,1H),1.20 ~1.54(m,6H),1.10(s,3H),1.07(m,1H),0.98 ~1.00(dd,J1=12.6 Hz,J2=2.4 Hz,1H),0.85 ~0.90(dt,J1=12.6 Hz,J2=3.6 Hz,1H),0.81(s,3H),0.80(s,3H),0.73(s,3H);IR(KBr)v/cm-1:3454,3130,3000,2931,2863,1681,1566,1469,1409,1387,755,708。
1.2.4 化合物1c的合成
在N2保护下,将1.00 g镁条加入到圆底烧瓶中,然后加入5 mL无水THF和少许碘粒。在室温下缓慢滴加2.2 mL对溴甲苯的THF(10 mL)溶液。滴加完毕后,加热到40℃反应1 h。将所制备的格式试剂冷却至0~5℃备用。将1.00 g(0.004 mol)香紫苏内酯的THF(10 mL)溶液滴加到上述格式试剂中。滴加完毕后,在0~5℃温度下反应15 min,加水淬灭反应,过滤,用THF洗涤固体,乙酸乙酯萃取,合并有机相并用无水硫酸镁干燥,过滤,旋蒸,经硅胶柱层析(V石油醚/V乙酸乙酯=10/1)得到白色固体1.19 g,产率 87%。mp:130 ~133 ℃;APCI-MS,m/z:342.2,[M - H]-,341.3;1H NMR(600 MHz,CDCl3):δ 7.92(d,J=7.8 Hz,2H),7.28(d,J=7.8 Hz,2H),3.12(dd,J1=18 Hz,J2=4.8 Hz,1H),2.97(dd,J1=17.4 Hz,J2=4.8 Hz,1H),δ 2.44(s,3H),2.23(t,J=4.8 Hz,1H),1.98 ~ 2.02(td,J1=12.6 Hz,J2=3.6 Hz,1H),1.71 ~ 1.76(br,d,J=13.8 Hz,1H),1.35 ~ 1.59(m,6H),1.20(s,3H),1.14 ~1.19(m,1H),1.00(dd,J1=12.6 Hz,J2=2.4Hz,1H),0.93 ~ 0.98(dt,J1=12.6 Hz,J2=3 Hz,1H),0.90(s,6H),0.82(s,3H);IR(KBr)v/cm-1:3486,2932,2866,1677,1606,1510,1458,1388,817。
1.2.5 化合物2a的合成
将0.5 g(1.5 mmol)化合物1a和3.5 mL 水合肼加入到10 mL一缩二乙二醇中。加热到120℃反应2 h,蒸出剩余的水合肼。再加入0.18 g(3.15 mmol)KOH,在200℃下反应至原料消失 (TLC)。冷却至室温,加入1N HCl(15 mL),乙醚萃取 (3×20 mL)。合并有机相,用饱和氯化钠溶液洗涤,干燥,旋蒸,经硅胶柱层析(V石油醚/V乙酸乙酯=10/1)得白色固体0.30 g,产率64%。mp:110~111℃;ESI-MS,m/z:314.2,[M - H]-,313.2;1H NMR(600 MHz,DMSO - d6):δ 7.25(t,J=7.2 Hz,2H),7.18(d,J=7.2 Hz,2H),7.14(t,J=7.2 Hz,1H),3.99(s,1H),2.70 ~ 2.75(td,J1=12.6Hz,J2=5.4 Hz,1H),2.50 ~ 2.55(td,J1=12.6 Hz,J2=5.4 Hz,1H),1.70 ~1.73(dt,J1=12.6 Hz,J2=3 Hz,1H),1.65 ~ 1.68(dt,J1=13.2 Hz,J2=3.6 Hz,2H),1.32 ~1.58(m,6H),1.18 ~ 1.25(ddd,J1=16.2 Hz,J2=13.2 Hz,J3=3 Hz,1H),1.10 ~ 1.14(m,2H),1.00(s,3H),0.90 ~ 0.95(td,J1=13.2 Hz,J2=3.6 Hz,1H),0.87 ~ 0.89(dd,J1=12 Hz,J2=1.8 Hz,1H),0.85(s,3H),0.76(s,3H),0.73(s,3H);IR(KBr)v/cm-1:3344,3060,2998,2923,2870,1605,1495,1454,1387,751,698。
1.2.6 化合物2b的合成
将0.5 g(1.6 mmol)化合物1b 和3.5 mL 水合肼加入到10 mL一缩二乙二醇中。加热到120℃反应2 h,蒸出剩余的水合肼。再加入0.19 g(3.36 mmol)KOH,在200℃下反应至原料消失 (TLC)。冷却至室温,加入1 N HCl(15 mL),乙醚萃取(3×20 mL)。合并有机相,用饱和氯化钠溶液洗涤,干燥,旋蒸,经硅胶柱层析 (V石油醚/V乙酸乙酯=10/1)得白色固体0.38 g,产率79%。mp:104 ℃;ESI-MS,m/z:304.2,[M - H]-,303.2;1H NMR(600 MHz,CDCl3):δ 7.30(s,1H),6.27(s,1H),6.01(d,J=3 Hz,1H),2.69 ~2.78(m,2H),1.85 ~1.88(dt,J1=12.6 Hz,J2=3 Hz,1H),1.35 ~1.79(m,9H),1.23 ~1.30(ddd,J1=15.5 Hz,J2=13.2 Hz,J3=3 Hz,1H),1.15(s,3H),1.11(t,J=4.2 Hz,1H),0.89 ~0.95(m,2H),0.87(s,3H),0.81(s,3H),0.79(s,3H);IR(KBr)v/cm-1:3470,2994,2954,2933,2858,1596,1508,1457,1388,736,722。
1.2.7 化合物2c的合成
将0.74 g(2.16 mmol)化合物1c和 5.2 mL 水合肼加入到20 mL一缩二乙二醇中。加热到120℃反应2 h,蒸出剩余的水合肼。再加入0.25 g(4.54 mmol)KOH,在200℃下反应至原料消失 (TLC)。冷却至室温,加入1 N HCl(25 mL),乙醚萃取(3×30 mL)。合并有机相,用饱和氯化钠溶液洗涤,干燥,旋蒸,经硅胶柱层析 (V石油醚/V乙酸乙酯=10/1)得白色固体0.54 g,产率76%。mp:124 ℃;ESI-MS,m/z:328.2,[M - H]-,327.1;1H NMR(600 MHz,DMSO - d6):δ 7.06(s,4H),3.96(s,1H),2.67(td,J1=12.6 Hz,J2=5.4 Hz,1H),2.47(td,J1=12.6 Hz,J2=5.4 Hz,1H),2.25(s,3H),1.69 ~1.72(dt,J1=12 Hz,J2=3Hz,1H),1.28 ~1.67(m,8H),1.18~1.25(ddd,J1=16.2 Hz,J2=13.8 Hz,J3=3 Hz,1H),1.09 ~1.14(m,2H),0.99(s,3H),0.91 ~ 0.96(td,J1=12.6 Hz,J2=3 Hz,1H),0.86 ~ 0.89(dd,J1=12.6 Hz,J2=2.4 Hz,1H),0.85(s,3H),0.76(s,3H),0.73(s,3H);IR(KBr)v/cm-1:3343,2998,2929,2870,1513,1458,1387,811。
2 结果与讨论
2.1 化合物1(a-c)的合成
化合物1a和1c通过格式试剂开环内酯环来合成。在格式试剂制备中,温度不宜过高,否者会发生格式试剂与卤代芳烃的偶联等副反应,导致产率降低,杂质增加。采用格式试剂开环内酯环时,延长反应时间或升高反应温度,都会产生少量含有两个苯环的化合物。实验中,采用较低的反应温度,通过TLC实时监测反应,待原料反应完全后及时猝灭反应,可以最大限度避免酮羰基进一步发生亲核加成反应。
合成化后物1b所需要的2-溴代呋喃时,要控制NBS的滴加速度,滴加过快,易增加双溴代呋喃的生成,实验中,我们加入了1倍当量和2倍当量的NBS,结果发现当加入1倍当量的NBS时,2-溴代呋喃的产率有所提高。本文中用参考文献[20]的方法来合成化合物1b。实验中,我们采取了两种加料方式,即将溴代呋喃加入到香紫苏内酯中反应或将香紫苏内酯加入到溴代呋喃中反应,后者加料方式较为方便,但是产率略有降低。
采用APCI或ESI负离子模式,化合物1(a-c)的液相-质谱数据表明所合成化合物的分子量正确,而且只引入了一个芳环。从IR数据来看,化合物1(a-c)均有明显的 -OH伸缩振动吸收峰(3340~3490 cm-1)以及酮C=O的伸缩振动吸收峰(1673~1681 cm-1)。IR数据也表明,通过格式试剂已经成功实现内酯的开环,得到含有一个芳环的Labdane二萜类似物。在1H NMR谱图中,1(a-c)化合物因为羰基的存在,呋喃环或苯环氢的化学位移均向低场移动,δ 7.3~8.0区间为苯环上氢的峰位置,δ 7.51,7.16 和6.46 为呋喃环上3 个氢的峰位置。
2.2 化合物2(a-c)的合成
化合物1(a-c)分子中均含有一个酮羰基,可以通过黄鸣龙、克莱门森等反应来还原为亚甲基。由于化合物1(a-c)分子中还存在一个叔羟基,在酸性条件下易于发生脱水、重排等副反应,因此选择碱性条件下的黄鸣龙反应来还原1(a-c)的羰基。黄鸣龙还原反应中,酮羰基首先与肼缩合得到腙类中间体,进一步还原得到对应的亚甲基。实验发现,化合物1(a-c)与水合肼反应时,以乙醇或一缩二乙二醇作为溶剂均可得到腙类化合物。考虑到第二步反应中也需要用到一缩二乙二醇溶剂,我们在腙的合成中也选用一缩二乙二醇作为溶剂,通过一锅煮的方法来还原羰基。这样无需乙醇去除步骤,反应时间较短,且产率有所提高。
采用APCI或ESI负离子模式,化合物2(a-c)的液相-质谱数据表明所合成化合物的分子量正确,羰基已被还原。从 IR数据来看,2(a-c)在1673~1681 cm-1处无酮C=O的伸缩振动吸收峰,也表明羰基已被还原。在1H NMR谱图中,化合物1(a-c)的羰基还原为亚甲基后,在δ 2.7附近有2个苄位氢的核磁信号,证明羰基已被还原。
3 结论
以香紫苏内酯为原料,通过内酯开环反应、黄鸣龙反应可以简便地合成Labdane型二萜类天然产物。该方法具有原料简单易得,步骤简短,后处理简单等优点,易于结构多样性地合成得到二萜“类天然产物”,为后期生物活性筛选等奠定基础。
[1]BURKE M D,BERGER E M,SCHREIBER S L.Generating diverse skeletons of small molecules comb-inatorially[J].Science,2003,302,613 -618.
[2]KOPP F,STRATTON C F,AKELLAa L B,et al.A diversity-oriented synthesis approach to macrocycles via oxidative ring expansion[J].Nat.Chem.Biol,2012,8(4):358-365.
[3]TAN D S.Diversity - oriented synthesis:exploring the intersections between chemistry and biology[J].Nat.Chem.Biol.2005,1(2):74-84.
[4]郭宗儒.天然产物的结构改造[J].化学进展,2012,47(2):144-157.
[5]周宇,张磊,栗增,等.高效合成技术在药物发中的应用[J].药学学报,2013,48(7):1014-1030.
[6]SCHREIBER S L.Target-oriented and diversity-ori ented organic synthesis in drug discovery[J].Science.2000,287:1964-1969.
[7]杨震.活性天然产物和结构多样性类天然产物的合成[J].化学进展,2009,21(1):47-54.
[8]刘钢,李裕林,南发俊.多样性导向的“类天然产物”化合物库合成[J].化学进展,2006,18(6):734-742.
[9]SHUL'TS E E,MIRONOV M E,KHARITONOV Y V.Furanoditerpenoids of the labdane seires:Occurrence in plants,total synthesis,several transformations and biological activity[J].Chem.Nat.Comp.2014,50(1):1-20.
[10]REUBEN J P.Two rings in them all:the labdanerelated diterpenoids[J].Nat.Prod.Rep.2010,27:1521 -1530,and previous reviews in this series.
[11]HERSEL U,STECK M,SEIFERT K.A new route to 2,7 - and 7 - functionalized labdanes[J].Eur.J.Org.Chem,2000,(8):1609-1615.
[12]KOLYMPADI M,LIAPIS M,RAGOUSSIS V.Synthesis of the marine furanoditerpene(- )- Marginatone[J].Tetrahedron,2005,61(8):2003-2010.
[13]ZORETIC P A,FANG H,RIBEIRO A A.Application of a radical methodology toward the synthesis of d,1 -5α -Pregnanes and related steroids:A stereoselective radical Cascade approach[J].J.Org.Chem.1998,63(21):7213-7217.
[14]JUSTICIA J,OLTRA J E,CUERVA J M.Palladium meddiated C-H activation in the field of terpenoids:synthesis of rostratone[J].Tetrahedron Lett,2004,45(22):4293-4296.
[15]JUSTICIA J,ROSALES A,BUNUEL E,el al.Titanocen ecatalyzed cascade cyclization of epoxypolyprenes:straightforward synthesis of terpenoids by free radical chemistry[J].Chem.Eur.J.2004,10(7):1778-1788.
[16]ZORETIC P A,FANG H.Synthesis of d,l-Norlabdane Oxide and Related Odorants:An Intramolecular Radical Approach[J].J.Org.Chem.1998,63(14):4779 -4785.
[17]NAKANO C,HOSHINO T,SATO T,et al.Substrate specificity of the CYC2 enzyme from Kitasato spora griseola:production of sclarene,biformene,and novel bicyclic diterpenes by the enzymatic reactions of labdaneand halimane - type diter pene diphosphates[J].Tetrahedron Lett,2010,51(1):125 -128.
[18]黄伟,王健波,唐功利.天然产物类药物的合成生物学研究[J].生命科学,2011,23(9):892-899.
[19]RAHEEM M A,NAGIREDDY J R,DURHAM R,et al.Efficient Procedure for the Preparation of 2-Bromofuran and Its Application in the Synthesis of 2 - Arylfurans[J].Syn.Comm.2010,40:2138-2146.
[20]MARGAROS I,VASSILIKOGIANNAKIS G.Synthesis of Chinensines A - E[J].J.Org.Chem.2007,72:4826-4831.
