阿瑞匹坦对癌症化疗患者口服羟考酮控释片药代动力学的影响
2015-07-07王绍志柴连海张勇邱志宏
王绍志,柴连海,张勇,邱志宏
(1.唐山市工人医院 药学部,河北 唐山 063500;2.唐山市工人医院 医学肿瘤科,河北 唐山 063500;3.河北省人民医院 药剂科,河北 石家庄 050051)
阿瑞匹坦对癌症化疗患者口服羟考酮控释片药代动力学的影响
王绍志1,柴连海2,张勇2,邱志宏3
(1.唐山市工人医院 药学部,河北 唐山 063500;2.唐山市工人医院 医学肿瘤科,河北 唐山 063500;3.河北省人民医院 药剂科,河北 石家庄 050051)
目的 探讨温和CYP3A4抑制剂阿瑞匹坦对口服羟考酮控释片药代动力学的影响。方法 本试验为单序列两期研究,其中癌痛患者每隔8 h或12 h连续多次口服羟考酮控释片。给药方案:第1~2天,单独予以羟考酮控释片;第3天,羟考酮控释片联合阿瑞匹坦(125 mg,早晨予以羟考酮的同时给药),给药8 h后测定羟考酮及其代谢产物的血药浓度,并测定第1~3天的稳态谷浓度(steady-state trough concentrations,Css)。结果 阿瑞匹坦使羟考酮的血药浓度-时间曲线下面积(AUC0-8)增大25%(P<0.001),使羟吗啡酮的AUC0-8增加34%(P<0.001),同时使去甲羟考酮的AUC0-8减少14%(P<0.001)。与第1天相比,第3天阿瑞匹坦使羟考酮的Css增加57%(P=0.001),使羟吗啡酮的Css增加36%(P<0.001),并使去甲羟考酮的Css减少24%(P=0.02)。结论 阿瑞匹坦临床用于多次服用羟考酮控释片治疗癌痛的患者可显著改变血药浓度水平,但无需改变羟考酮控释片的剂量。
阿瑞匹坦;羟考酮;药代动力学
羟考酮是一种广泛用于治疗癌痛和慢性疼痛的μ阿片受体激动剂[1]。大剂量或过量使用时,羟考酮可引起浅度呼吸抑制、嗜睡、昏睡或昏迷、骨骼肌松弛。阿瑞匹坦是可以口服的选择性神经激肽1受体激动剂,对化疗诱发的急性和延迟性恶心、呕吐有效,阿瑞匹坦经CYP同工酶1A2、2C19和3A4代谢,是CYP3A4中度抑制剂和CYP2C19、CYP2C9的极弱抑制剂[2]。羟考酮与阿瑞匹坦联合用药临床上可用于癌症患者护理。然而,阿瑞匹坦可能会通过抑制CYP3A介导的代谢来增加羟考酮与其代谢产物血药浓度。因此,羟考酮的副作用可能会增加。故本试验探讨了温和CYP3A4抑制剂阿瑞匹坦对癌痛患者口服羟考酮控释片药代动力学的潜在影响,现报道如下。
1 资料与方法
1.1 一般资料 患者为2013年1月~2014年12月在唐山市工人医院治疗的60例恶性肿瘤患者。纳入标准:①每天持续分2~3次服用羟考酮控释片治疗癌痛,并拟用阿瑞匹坦治疗化疗恶心呕吐(chemotherapy-induced nausea and vomiting,CINV)的化疗患者;②患者年龄≥18岁;③患者组织学或病理学确诊为恶性实体瘤,器官功能完善,血清总胆红素低于1.5倍正常高限值(upper limits of normal,ULN),天冬氨酸转氨酶低于2.5倍ULN,血清肌酐低于1.5倍ULN。排除标准:排除可能影响消化、影响羟考酮控释片或阿瑞匹坦吸收的肠胃疾病患者;排除正在接受或可能接受可作为强效CYP3A4或CYP2D6抑制剂或诱导剂的药物或食物的患者。本试验得到我院伦理委员会批准,所有患者或家属均签署了知情同意书。
1.2 仪器与试剂 液相色谱仪器(LC7300,济南海能仪器股份有限公司);质谱仪器(API400,美国AB公司);乙腈(德国MERCK公司);超纯水(美国Millipore公司)。
1.3 方法
1.3.1 用药方法:本试验是单序列两期研究。每组患者每隔8 h或12 h口服不同剂量羟考酮控释片(5、10、15、20 mg)(批准文号:国药准字J20040099,BARD PHARMACEUTICALS LIMITED生产)。第1~2天(化疗前1~2天),单独予以患者羟考酮控释片(Ⅰ期),第3天(化疗当日早晨),在化疗前1 h同时口服羟考酮控释片和阿瑞匹坦125 mg(批准文号:注册证号H20130545,Merck Sharp & Dohme Corp生产)(Ⅱ期)。患者按其肿瘤分型的标准治疗方案给予抗癌药,并在适当情况下使用地塞米松和5-羟色胺3(5HT3)进行止吐治疗。试验的主要终点是阿瑞匹坦给药后羟考酮及其代谢产物的药代动力学。次要终点是阿瑞匹坦给药后安全性及不良事件,包括恶心、呕吐、便秘和嗜睡等。研究期间全程记录患者特征和用药信息。采用CTCAE 4.0软件对不良事件进行评估。
1.3.2 血药浓度测定:羟考酮给药前、给药1、2、3、5和8 h后采集患者血样进行药代动力学分析。血液标本采集到肝素锂抗凝管0.5 h内,4 ℃条件下1500 g离心10 min,进行血浆分离,并于-80 ℃下保存备用。采用高效液相色谱串联质谱法测定羟考酮、去甲羟考酮和羟吗啡酮的血药浓度,采用纯乙腈超声提取,高速离心沉淀蛋白后,以乙腈-乙酸铵缓冲液作为流动相,100A反相色谱柱,在正离子模式下以电喷雾电串联质谱进行测定。
1.3.3 药代动力学分析:用Phoenix WinNonlin 6.0软件进行羟考酮、去甲羟考酮和羟吗啡酮的药代动力学分析。观察并记录最高血药浓度(Cmax)、血药浓度到达峰值的时间(Tmax)与稳态谷浓度(Css)。用梯形法计算0~8 h的药时曲线下面积(AUC0-8)。用对数线性梯形法计算血药浓度增加值与降低值。

2 结果
2.1 患者一般资料 20例患者经评估符合入选标准。患者特征见表1。男性17例,女性3例,美国东部肿瘤协作组体力状态评分(Eastern Cooperative Oncology Group Performance Status,ECOG)为1~2分。主要肿瘤分型为胰腺癌和头颈癌,所有患者均为Ⅳ期。各患者每隔8 h或12 h定期口服适当剂量的羟考酮控释片,根据患者肿瘤类型及患病程度选择合适的用药剂量,根据用药剂量对患者进行分组,羟考酮平均日剂量为20 mg(10~60 mg),平均21.5 mg。见表2。

表1 患者的一般特征Tab.1 General characteristics of patients

表2 羟考酮剂量Tab.2 Oxycodone dose
2.2 羟考酮及其代谢产物的药代动力学 首先对所有20例患者羟考酮及其代谢产物的药代动力学进行评估。患者分为4组,分别口服5、10、15、20 mg羟考酮;每组中又分为8 h或12 h服用2亚组(表2),其中每12 h口服5 mg羟考酮控释片的患者中,羟吗啡酮血药浓度在定量下限以下。表3和表4概括了羟考酮单独给药或羟考酮联合阿瑞匹坦的药代动力学参数,图1显示每隔12 h单独予以10 mg羟考酮控释片或羟考酮联合阿瑞匹坦的患者血药浓度的几何平均值,羟考酮控释片联合阿瑞匹坦(1102 ng·h/mL,CV29.9%)与单用羟考酮控释片的羟考酮AUC0-8几何平均值(882 ng·h/mL,CV35.7%),比值为1.25(95%CI:1.14~1.36;CV21.8%;P=0.00004),羟考酮控释片联合阿瑞匹坦(2.79 ng/mL,CV28.0%)与单用羟考酮控释片的羟考酮Cmax(2.28/mL,CV31.4%) 比值为1.22(95%CI:1.11~1.34;CV20.6%;P=0.0002)。去甲羟考酮联合阿瑞匹坦(616 ng·h/mL,CV51.6%)与单用甲羟考酮的去甲羟考酮AUC0-8几何平均值 (718 ng·h/mL,CV45.2%)比值为0.86(95%CI:0.81~0.91;P=0.00005),羟吗啡酮联合阿瑞匹坦(20.7 ng·h/mL,CV65.8%)与单用羟考酮控释片的羟吗啡酮AUC0-8几何平均值(14.9 ng·h/mL,CV78.0%)比值为1.34(95%CI:1.20~1.49;P=0.00004)。羟考酮及其代谢产物的血药浓度明显受阿瑞匹坦的存在与否影响。
由于达到稳态,故第1天羟考酮及其代谢产物的谷浓度与第2天羟考酮及其代谢产物的谷浓度相似。第3天联合阿瑞匹坦后谷浓度较第1、2天高。对于羟考酮、去甲羟考酮和羟吗啡酮,第3天羟考酮控释片联合阿瑞匹坦的几何平均谷浓度与第2天单用羟考酮控释片的几何平均谷浓度之比分别为1.65(P=0.0001)、0.796(P=0.00001)和1.32(P=0.02)。见表4。
在本研究和临床实践中,羟考酮与合用阿瑞匹坦后,患者的不良反应和副作用均无提升。

图1 每隔12 h服用10 mg羟考酮控释片患者与联合阿瑞匹坦的患者的羟考酮、去甲羟考酮和羟吗啡酮平均血药浓度曲线Fig.1 Oxycodone, oxycodone and oxymorphone mean plasma concentration curve when every 12 h after taking 10 mg of oxycodone controlled release tablets combined with aprepitant

项目羟考酮去甲羟考酮羟吗啡酮Cmax(ng/mL)Tmax(hr)AUC0-8(ng·h/mL)AUC0-8(ng·h/mL)AUC0-8(ng·h/mL)患者数2020202015*未用阿瑞匹坦2.28±0.172.67±0.21882±58718±9214.9±1.48CV-31.40%-57.70%-35.70%-45.20%-78.00%使用阿瑞匹坦2.79±0.143.62±0.281102±82616±4120.7±2.04CV-28.00%-32.10%-29.90%-51.60%-65.80%比值,均值(范围)1.22(1.11~1.34)1.25(1.14~1.36)0.86(0.81~0.91)1.34(1.20~1.49)P0.00020.070.000040.000050.00004
AUC0-8:0~8 h的药时曲线下面积;比值:羟考酮联合阿瑞匹坦与单药羟考酮的几何平均值之比。*:5例因低于定量下限而排除。
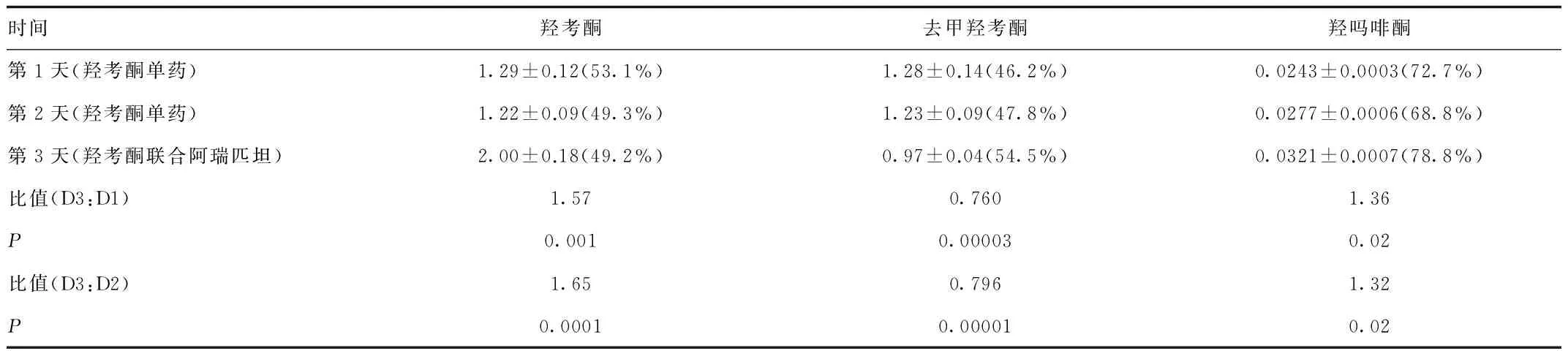
表4 羟考酮及其代谢产物的谷浓度 (ng/mL)Tab.4 Minimum concentration of oxycodone and its metabolites (ng/mL)
比值(D3:D1),第3天羟考酮控释片联合阿瑞匹坦的几何平均谷浓度与第1天单用羟考酮控释片的几何平均谷浓度之比;比值(D3:D2),第3天羟考酮控释片联合阿瑞匹坦的几何平均谷浓度与第2天单用羟考酮控释片的几何平均谷浓度之比
3 讨论
羟考酮是一种吗啡半合成衍生物,具有与吗啡类似的镇痛作用和副作用[3]。羟考酮主要经十二指肠和肝细胞色素P450(CYP)的同工酶代谢[4-5]。羟考酮主要代谢途径是CYP3A4介导N-去甲基作用生成去甲羟考酮,同时少部分通过CYP2D6介导3-O-去甲基作用生成活性代谢产物羟吗啡酮,这些代谢产物经CYP2D6和CYP3A4进一步氧化生成去甲羟吗啡[4]。
阿瑞匹坦可与5HT3拮抗剂和糖皮质激素(如地塞米松)联合用于高中度致吐性化疗。阿瑞匹坦经CYP同工酶1A2、2C19和3A4代谢,是CYP3A4中度抑制剂和CYP2C19与CYP2C9的极弱抑制剂[2]。研究表明阿瑞匹坦可抑制CYP3A4酶活性[6]。在口服阿瑞匹坦治疗CINV所用的125 mg/80 mg方案中,第1、3天血药峰浓度(Cmax)约在4 h(Tmax)分别达到1600 μg/mL和1400 μg/mL。由于口服阿瑞匹坦后的肠道药物浓度比血药浓度高,因此口服阿瑞匹坦比静注阿瑞匹坦更能抑制肠道CYP3A4,且口服联合用药受肠道CYP3A4抑制作用的程度比静注联合用药更大[6-7]。
本试验表明阿瑞匹坦抑制CYP3A4介导N-去甲基作用可使羟考酮的AUC增加25%,使去甲羟考酮的AUC减少14%,同时通过改变CYP2D6途径将羟吗啡酮的AUC连续增加34%。预先估计羟考酮与阿瑞匹坦之间的相互作用有临床意义显著性,AUC0-8比值增加33%(CV45.1%)。在本试验中,由于合用阿瑞匹坦,羟考酮的不良反应和副作用均无提升。几何平均AUC08之比增大25%(中值1.25;95%CI:1.14~1.36)具有统计学意义,其程度低于预期,此时无需在临床上对癌症患者使用阿瑞匹坦时改变羟考酮控释片的剂量。活性代谢产物羟吗啡酮是一种较羟考酮μ阿片受体亲和力低4~6倍,且浓度也比其低的强效阿片样物质[4,8],故羟吗啡酮不可能对临床意义有显著影响。但由于阿瑞匹坦3 d的推荐剂量为125 mg/80 mg,故应进一步探讨125 mg/80 mg阿瑞匹坦方案对癌痛患者口服羟考酮控释片药代动力学的潜在影响。
由于阿瑞匹坦肠道浓度较血药浓度高,口服联合用药受肠道CYP3A4抑制作用影响的程度较静注给药大。因此,本试验这一结果可能对静注羟考酮不适用。虽然给药前1天的羟考酮及其代谢产物的谷浓度与给药前2天的羟考酮及其代谢产物的谷浓度相似,但第3天联合阿瑞匹坦后的这些谷浓度均高于第1、2天。这表明稳态时单用羟考酮控释片的谷浓度未见日间变异性(见表4)。同时,羟考酮控释片联合阿瑞匹坦与单用羟考酮控释片的几何平均AUC0-8和谷浓度之比分别为1.25(0.98~1.96)和1.65(0.54~3.41),个体间变异系数较广。
综上所述,阿瑞匹坦因抑制CYP3A4介导的N-去甲基作用使羟考酮药量增加25%。多剂量服用羟考酮控释片治疗癌痛的患者临床使用阿瑞匹坦可显著改变血药浓度水平,但无需在癌痛患者临床合用阿瑞匹坦过程中改变羟考酮控释片的剂量。
[1] 王特, 朱瑞新, 田川, 等. 具有μ/δ双重功效的阿片受体激动剂中药成分筛选研究[J]. 辽宁中医药大学学报, 2014, 16(2):46-51.
[2] 史鹤玲, 李雪冰, 张同梅, 等. 癌痛患者疼痛门诊药物治疗的临床效果分析[J]. 中国肿瘤临床, 2013,40(24):1506-1509.
[3] Kawakami J.Clinical pharmacology and pharmacoepidemiology for medication safety in clinical settings[J].Yakugaku Zasshi, 2015,135(4):619-624.
[4] Goncalves L, Friend LV, Dickenson AH.The influence of μ-opioid and noradrenaline reuptake inhibition in the modulation of pain responsive neurones in the central amygdala by tapentadol in rats with neuropathy[J]. Eur J Pharmacol, 2015(749):151-160.
[5] Panebianco D, Dean D, Kraft WK,et al. Double-blind crossover study to assess potential differences in cytochrome P450 3A4 activity in healthy subjects receiving ondansetron plus dexamethasone, with and without aprepitant[J].Cancer Chemother Pharmacol,2011, 67(6):1313-1321.
[6] McCrea JB, Majumdar AK, Goldberg MR, et al. Effects of the neurokinin1 receptor antagonist aprepitant on the pharmacokinetics of dexamethasone and methylprednisolone[J]. Clin Pharmacol Ther, 2003, 74(1): 17-24.
[7] Nygren P, Hande K, Petty KJ, et al. Lack of effect of aprepitant on the pharmacokinetics of docetaxel in cancer patients[J]. Cancer Chemother Pharmacol, 2005, 55(6): 609-616.
[8] Loos WJ, de Wit R, Freedman SJ, et al. Aprepitant when added to a standard antiemetic regimen consisting of ondansetron and dexamethasone does not affect vinorelbine pharmacokinetics in cancer patients[J]. Cancer Chemother Pharmacol, 2007, 59(3): 407-412.
[9] Majumdar AK, McCrea JB, Panebianco DL, et al. Effects of aprepitant on cytochrome P450 3A4 activity using midazolam as a probe[J]. Clin Pharmacol Ther, 2003, 74(2): 150-156.
[10] Pergolizzi JV, Köknel Talu G, Zmponga G, et al. Maximizing value in opioid utilization: Is oxycodone immediate release a good option for pain management? [J]. Agri, 2015, 27(1):1-11.
(编校:谭玲)
Effect of aprepitant on pharmacokinetics of cancer chemotherapy patients with oral oxycodone controlled release tablets
WANG Shao-zhi1,CHAI Lian-hai2, ZHANG Yong2, QIU Zhi-hong3
(1.Department of Pharmacy,Tangshan Worker’s Hospital,Tangshan 063500,China;2.Department of Oncology,Tangshan Worker’s Hospital,Tangshan 063500,China;3 Department of Pharmacy, Hebei People’s Hospital, Shijiazhuang 050051,China)
ObjectiveTo investigate effect of the mild CYP3A4 inhibitor aprepitant on the pharmacokinetics of orally administered controlled-release (CR) oxycodone. MethodsThis study designed was an single-sequence with two phases in cancer patients with pain who continued to be administered orally with multiple doses of CR oxycodone every 8 or 12 hours. Plasma concentration of oxycodone and its metabolites were measured up to 8 hours after administration as follows: on day 1, CR oxycodone was administered alone; on day 2, CR oxycodone was administered with aprepitant (125 mg, at the same time of oxycodone dosing in the morning). The steady-state trough concentrations (Css) were measured from day 1 to day 3. ResultsAprepitant increased the area under the plasma concentration-time curve (AUC0-8) of oxycodone by 25% (P<0.001) and of oxymorphone by 34% (P<0.001), as well as decreased the AUC0-8 of noroxycodone by 14% (P<0.001). Moreover, aprepitant increased Css of oxycodone by 57% (P=0.001) and of oxymorphone by 36% (P<0.001) and decreased Css of noroxycodone by 24% (P=0.02) at day 3 compared to day 1. ConclusionThe clinical use of aprepitant in patients receiving multiple doses of CR oxycodone for cancer pain significantly altered plasma concentration levels, but would not appear to need modification of the CR oxycodone dose.
aprepitant;oxycodone;pharmacokinetics
王绍志,男,硕士,主管药师,研究方向:药理学,E-mail:wangshaozhits@163.com。
R441.1
A
1005-1678(2015)06-0133-04
