Curing Mechanism of Condensed Polynuclear Aromatic Resin and Thermal Stability of Cured Resin
2015-06-22
(College of Chemical Engineering, State Key Laboratory of Heavy Oil Processing, China University of Petroleum, Qingdao 266580)
Curing Mechanism of Condensed Polynuclear Aromatic Resin and Thermal Stability of Cured Resin
Li Shibin; Sun Qiqian; Wang Yuwei; Wu Mingbo; Zhang Zailong
(College of Chemical Engineering, State Key Laboratory of Heavy Oil Processing, China University of Petroleum, Qingdao 266580)
In order to improve the thermal stability of condensed polynuclear aromatic (COPNA) resin synthesized from vacuum residue, 1,4-benzenedimethanol was added to cure COPNA resin. The curing mechanism was investigated by proton nuclear magnetic resonance spectrometry, solid carbon-13 nuclear magnetic resonance spectrometry and Fourier transform infrared spectroscopy. Microstructures of the uncured and the cured COPNA resins were studied by scanning electron microscopy and X-ray diffractometry. The thermal stability of COPNA resins before and after curing was tested by thermogravimetric analysis. The element composition of the cured COPNA resin heated at different temperatures was analyzed by an element analyzer. The results showed that the uncured COPNA resin reacted with the cross-linking agent during the curing process, and the curing mechanism was confirmed to be the electrophilic substitution reaction. Compared with the uncured COPNA resin, the cured COPNA resin had a smooth surface, well-ordered and streamlined sheet structure with more crystalline solids, better molecular arrangement and orientation. The weight loss process of the uncured and cured COPNA resins was divided into three stages. Carbon residue of the cured COPNA resin was 41.65% at 600 ℃, which was much higher than 25.02% of the uncured COPNA resin, which indicated that the cured COPNA resin had higher thermal stability.
condensed polynuclear aromatic resin, synthesis, vacuum residue, curing, thermal stability
1 Introduction
The condensed ploynuclear aromatic (COPNA) resins have attracted considerable attention for their potential application as novel heat-resistant materials thanks to their good lubricity, mechanical properties and mouldability. The COPNA resins with a three-dimensional network structure were initially prepared by Otani S. in the 1980s[1]. In the earlier studies, pure polycyclic aromatics, including phenanthrene, naphthalene, anthracene, and pyrene, were used as monomers[2-4], resulting in a high cost of COPNA resins. Recently, some cheap materials with high aromatic content, such as coal tar pitch, petroleum pitch, ethylene tar, FCC slurry oil, and vacuum residue have been successfully used to prepare the COPNA resins[5-9].
Most of the relevant research works have been focused on the reaction mechanism and the properties of the uncured resins synthesized from different raw materials. There were few reports focusing on the curing mechanism and thermal behavior of the cured resins[10], especially for the resins synthesized from complicated materials. The heat resistance of the cured resins synthesized from rich aromatic materials was not significantly higher than the uncured resins according to our preliminary studies[8-9].
In this work, we attempt to improve the heat resistance of the cured COPNA resins by adding the curing agent into the uncured COPNA resin produced from vacuum residue. The curing mechanism, microstructure and thermal stability of the COPNA resins before and after curing were investigated.
2 Experimental
2.1 Raw materials
The COPNA resin was synthesized in the laboratory according to the procedures described in the literature[9]. The reagent 1,4-benzenedimethanol (A.R.) used as the curing agent was purchased from the Sinopharm Chemical Re-agent Company, China. Chloroform (A.R.) was obtained from the Xilong Chemical Co., Ltd., China.
2.2 Preparation of cured COPNA resin
Firstly, the COPNA resin was dissolved in chloroform, and then 1,4-benzenedimethanol was added into the solution according to a certain ratio. The cured COPNA resin was obtained after the solvent was evaporated and the residue was heated at 200 ℃ for 2 h in a nitrogen flow of 50 mL/min.
2.3 Characterization
The uncured and the cured COPNA resin samples were characterized by the proton nuclear magnetic resonance spectrometry (1H-NMR), the solid carbon-13 nuclear magnetic resonance spectrometry (13C-NMR) and the Fourier transform infrared spectroscopy (FT-IR). The surface texture and the heat resistance of the resins were investigated by the scanning electron microscopy (SEM) and the thermogravimetric analysis (TGA). The state of hydrogen atoms in the resins was characterized by a Bruker AV5001H-NMR spectrometer, using TMS as the internal standard and CDCl3as the solvent. The sweep length was 10 000 Hz, and the resonance frequency was 500 MHz. The solid carbon-13 NMR (CP TOSS13CNMR) spectra were collected using a Bruker Avance II 400 spectrometer operating at 100 MHz for13C. The rotor spin speed was 5 000 Hz. For all samples, the optimum relaxation delay (D1) and the contact time were 4 s and 30 ms, respectively. The FT-IR spectra of the uncured and the cured COPNA resins were recorded on a Shimadzu 8400S FT-IR spectrometer in the transmittance mode. The scan frequency of each spectrum was 15 s-1. The scanning electron microscopy (SEM) was carried out using a Philips XL30 environmental scanning electron microscope. The accelerated voltage was 20 kV, and the samples were coated with aurum in vacuum. Thermal properties of the resins were studied by a Shimadzu DTG-60 thermogravimetric analyzer at a heating rate of 10 ℃/min under a nitrogen flow of 50 mL/min. The X-ray diffraction (XRD) analyses were carried out with a Rigaku Miniflex instrument operating at a tube voltage of 40 kV and a tube current of 40 mA. The CuKα (λ=1.5405 Å) radiation was selected as the monochromic energy radiation. Elemental analysis was performed on a Vario EL organic element analyzer. The element contents of C, H and O in the cured COPNA resins that were heated at different temperatures were analyzed.
3 Results and Discussion
3.11H-NMR analysis
The1H-NMR spectra can reveal the distribution of protons in different chemical environments. Figure 1 displays the1H-NMR spectra of the uncured and the cured COPNA resins. Table 1 shows the contents of the different hydrogen atoms observed by1H-NMR spectra based on the peak areas, where HA, HA1, HA2and HA3refer to the aromatic hydrogen and the hydrogen attached to the simple cyclic aromatics, the double-cyclic aromatics and the three-cyclic aromatics, respectively. Hα1and Hα2denote the hydrogen in groups of α-CH3, α-CH2and α-CH that are connected with one aromatic ring and two aromatic rings, respectively. Similarly, Hβand Hγrefer to the hydrogen in β-CH2, β-CH, and γ-CH3that is connected with the aromatic ring[11-13]. These differences in hydrogen atom content can be used to analyze the changes of hydrogen atoms during the curing process, which is beneficial to the explanation of the mechanism for curing of the COPNA resins. Table 1 shows that the HA1content of the cured COPNA resins is slightly higher than that of the uncured COPNA resin. This might be caused by 1,4-benzenedimethanol, which had a number of aromatic hydrogen atoms attached to simple cyclic aromatic ring. In addition, it was noteworthy that, after curing, the contents of HA, HA2, HA3and Hα1reduced from 43.02%, 16.82%, 10.01% and 28.01% to 34.70%, 10.87%, 4.92% and 18.41%, respectively. At the same time, the content of Hα2increased from 8.00% ofthe uncured COPNA resin to 21.84% of the cured resin. The above results indicated that during the curing process some methylene groups attached to one aromatic ring reacted with another aromatic hydrocarbons, and meanwhile, some hydrogen atoms attached to the aromatic ring were replaced.

Figure 11H-NMR spectra of uncured and cured COPNA resins
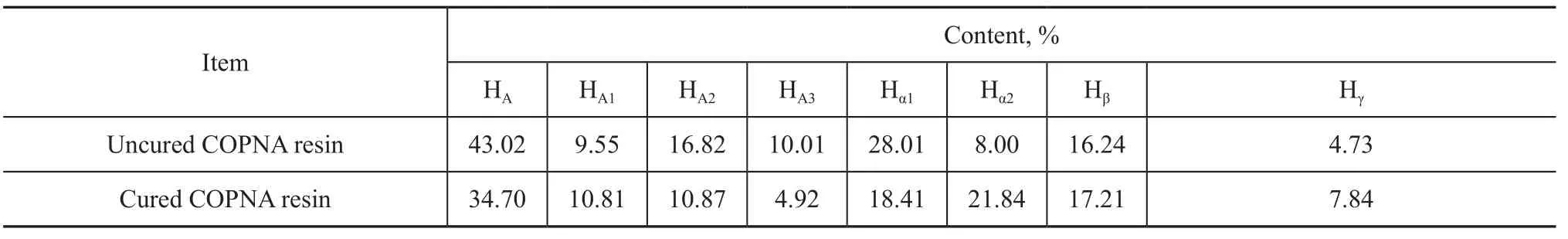
Table 1 Contents of different H bands in uncured and cured COPNA resins
3.2 Solid-state13C-NMR analysis
According to the literature[11,13-17], aliphatic carbon atoms (CS) and aromatic carbon atoms (CA) can be detected by the13C-NMR spectrometry. Moreover, aliphatic carbon atoms can be divided into CS1, CS2and CS3, and aromatic carbon atoms can be divided into CAH, CANRand CAO. The classification and results of carbon distribution of carbon types are given in Table 2.Figure 2 presents the solid-state13C-NMR spectra of the uncured and the cured COPNA resins. Solid-state13CNMR peak areas are a semi-quantitative measure of the relative contributions of different functional groups to the organic carbon that is present in a sample. Although the carbon peak areas cannot be used to accurately estimate the contents of each kind of carbon functional groups, but the ratio of carbon peak areas can reflect the relative change of various carbon atoms in the COPNA resin before and after curing. The area ratio of the peaks at chemical shift (δ) of 100—130 to that of 130—150 (S100-130/ S130-150) reflects the relative amount of CAHto CARN. Similarly, the area ratio of the peaks at δ of 32—55 to δ of 0—32 (S32-55/S0-32) reflects the relative amount of CS3to Cs1and CS2, respectively. Specifically, the S100-130/S130-150ratio of the uncured and the cured COPNA resins was found to be 1.376 2 and 1.233 7, respectively, whereas the S32-55/S0-32ratio of the uncured and the cured COPNA resin was 0.621 8 and 0.752 3, respectively. These changes indicated that the electrophilic substitution reaction of aromatic hydrocarbon occurred during the curing process, resulting in not only the replacement of aromatic hydrogen, but also the decrease of CAH, and the increase of CARNand CH2-α attached to aromatic ring. The results were in agreement with the1H-NMR analysis.

Table 2 Assignment for13C-NMR chemical shifts

Figure 213C-NMR spectra of uncured and cured COPNA resins
3.3 FT-IR analysis
The FT-IR spectra of the uncured and the cured COPNA resins are shown in Figure 3. The peak near the wavenumber of 3 045 cm-1was ascribed to stretching vibrations of aromatic C-H bonds. The peak near 1 460 cm-lwas attributed to the bending stretching of CH3and CH2groups. The peak near 1 380 cm-lwas related to the asym-metrical deformation vibration of CH3groups. These peaks between 900 cm-1and 600 cm-1were caused by the out-of-plane vibration of aromatic C-H bonds. The peak near 1 600 cm-1corresponded to aromatic C=C bond[18-19]. Many researchers[20-21]have reported a linear relationship between the absorbance ratio of bands in the range from 1 460 cm-1to 1 380 cm-1(A1460/A1380) and the molar ratio of methylene to methyl groups (n(CH2) /n(CH3)) in molecules. Therefore, the trend for change of methylene group relative to methyl group during the curing process can be found by comparing the A1460/A1380values of the uncured and the cured COPNA resins. The A1460/A1380value increased from 1.277 1 of the uncured COPNA resin to 1.337 7 of the cured COPNA resin, which indicated that the n(CH2) /n(CH3) value increased during the curing process of the resins. We believe that the addition of curing agent, which contains a number of methylene groups, contributes to the changes. The area ratio of the peaks at 670—900 cm-1and 1600 cm-1(S670-900/S1600) can be used to characterize the substitution degree of hydrogen atoms attached to aromatic rings. The lower the S670-900/S1600value is, the more the hydrogen atoms that are attached to aromatic rings are substituted. The S670-900/S1600values reduced from 3.339 2 of the uncured COPNA resin to 2.019 5 of the cured resin, which indicated that some aromatic hydrogen atoms were substituted and the degree of cross-linking increased during the curing process. This analysis was consistent with the1H-NMR and13C-NMR analyses.
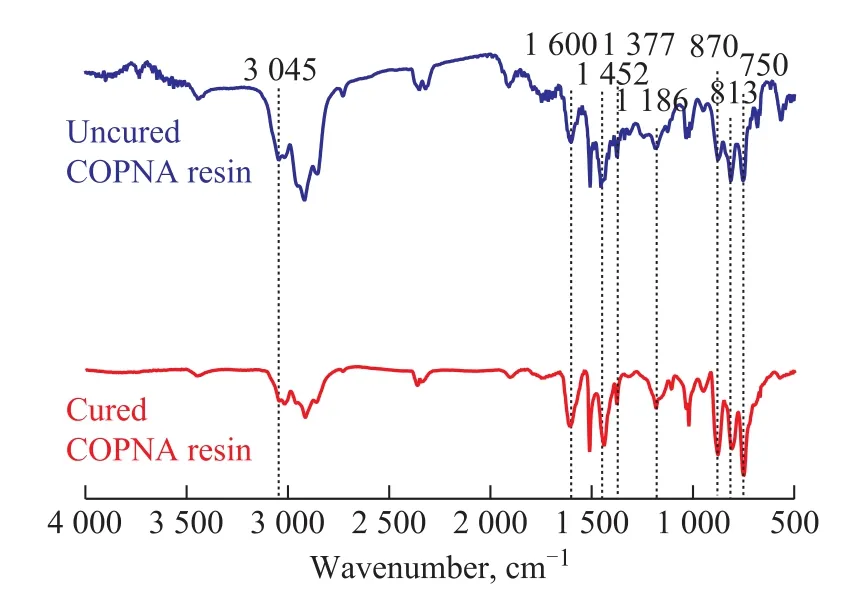
Figure 3 FT-IR spectra of uncured and cured COPNA resins
3.4 Curing mechanism of COPNA resin
Based on the results of1H-NMR,13C-NMR and FT-IR spectrometric analyses, it can be concluded that the electrophilic substitution reaction occurs between the uncured COPNA resin and the curing agent. Firstly, the alcoholic hydroxyl groups of 1,4-benzenedimethanol react on protons under the acidic condition to give benzyl carbonium ions, and then the electrophilic substitution reaction occurs via subsequent attack of the benzyl carbonium ions on aromatic rings of COPNA resin. Thus, macromolecular compound with a three-dimensional network structure is formed. The curing mechanism and the molecular structure model of the resins are shown in Figure 4.

Figure 4 The curing mechanism of COPNA resins
3.5 SEM analysis
Figure 5 presents the SEM images of the uncured and the cured COPNA resins. The photographs of smaller magnification exhibited that the uncured and the cured COPNA resins were both of irregular blocky shape. A larger magni fication revealed that the uncured COPNA resin with a rough surface and cross-section had no apparent orientation texture, thereby indicating that the uncured COPNA resin was isotropic. However, the cured COPNA resin displayed a well-ordered streamlined sheet structure. This might be due to the occurrence of the cross-linking reaction between the condensed aromatic sheet and 1,4-benzenedimethanol during the curing process, which could lead to a better molecular arrangement and orientation of the cured COPNA resin.
3.6 XRD analysis

Figure 5 SEM images of uncured (a) and cured (b) COPNA resins
The XRD patterns of the uncured and the cured COPNA resins are shown in Figure 6. It can be seen from Figure 6 that both the uncured and the cured COPNA resins represented a broad band between 10° and 30°, indicating that the resins were amorphous in structure generally. The XRD parameters of the resins are given in Table 3. The two weak peaks of uncured COPNA resin at 21.48° and 23.72° were assigned to the γ-band, which is thought to be the packing distance of saturated structures (aliphatic chains or condensed saturated rings)[23], while the cured COPNA resin exhibited eight weak peaks. The five former peaks were classified to be the γ-peak, and two other peaks centered at 25.36° and 43.68° were considered to be (002) and (100) diffraction of the graphite[24]. All these data indicated that more crystalline solid appeared while the condensation degree of COPNA resin was increased, and a graphite-like carbon material was generated during the curing process. It can be concluded that the cured COPNA resin has a better molecular arrangement and orientation than the uncured resin just as mentioned in Section 3.5.

Figure 6 XRD patterns of uncured and cured COPNA resins

Table 3 XRD parameters of uncured and cured COPNA resins
3.7 Thermal behavior analysis

Figure 7 TG-DTG curves of uncured COPNA resin
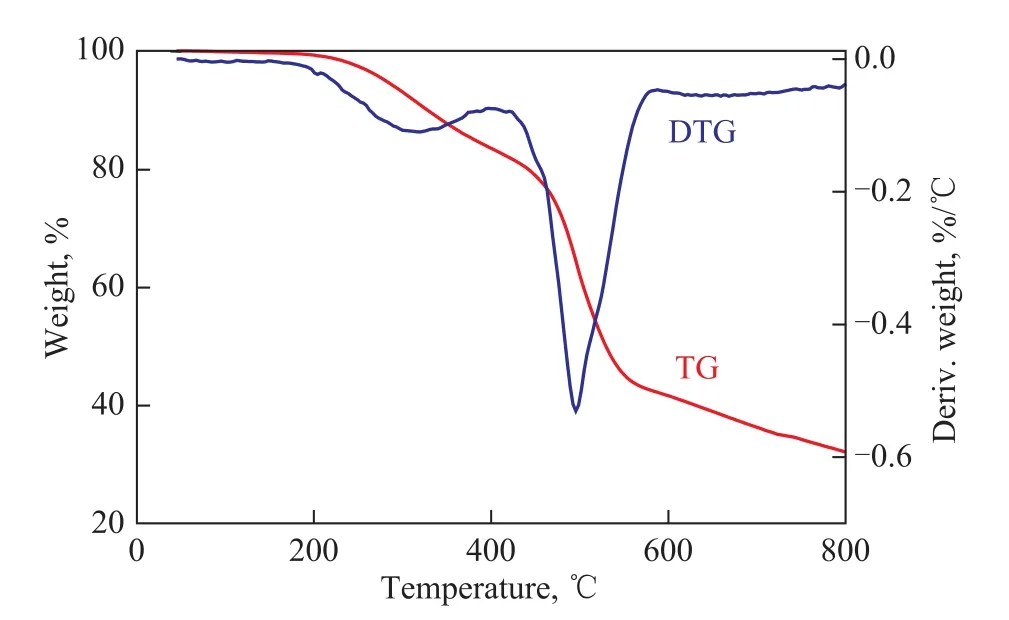
Figure 8 TG-DTG curves of cured COPNA resin
The TG-DTG curves of the uncured and the cured COPNA resins are shown in Figures 7 and 8. The TGDTG curves illustrated that the weight loss of the uncuredand the cured COPNA resins could be divided into three stages. Based on the first weight loss stage in TG-DTG curves, we can confirm that the temperature acting on the uncured and the cured COPNA resins ranged from 230 ℃ to 350 ℃ and from 280 ℃ to 460 ℃, respectively, in which the weight loss was mainly ascribed to the evaporation of water and uncrosslinked low molecular weight compounds. In the second weight loss stage, the temperature acting on the uncured and the cured COPNA resins ranged from 350 ℃ to 520 ℃ and 460 ℃ to 580 ℃, respectively, in which the weight loss was mainly caused by the removal of light molecules generated from the multiple and complex reactions (cracking, polymerization, condensation, etc.). In the third stage, the resins were already transformed into a semi-coke solid state. In this interval, small molecules, such as H2and CH4, were evolved from the cyclization and aromatization reactions[24-27].
The DTG curve of the uncured COPNA resin showed that there were four maximum weight loss rate (Tmax) peaks appearing at 244 ℃, 336 ℃, 443 ℃ and 465 ℃, respectively. However, in the case of DTG analysis of the cured COPNA resin, only two Tmaxpeaks appeared at 317 ℃ and 495 ℃. This shows that thermal decomposition mechanisms of the uncured and the cured COPNA resins are slightly different.
Table 4 shows the thermogravimetric parameters of the uncured and the cured COPNA resins. It can be observed that all the temperatures relating to the initial weight loss (Ti), the maximum weight loss rate (Tmax) and the final weight loss (Tf) of the cured COPNA resin were much higher than those of the uncured COPNA resin. Tiis one of the main criteria for heat stability of resins. The higher the value of Ti, the higher the thermal stabilityis. The weight loss rate of the cured COPNA resin was only 17.41% at 400 ℃, which was remarkably lower as compared to that of the uncured resin (45.28%). Although a lot of light molecules generated from the cracking, polymerization and condensation reactions were removed, the carbon residue of the cured COPNA resin still remained at 41.65% at 600 ℃, which was much higher than that of the uncured COPNA resin (25.02%). The results mentioned above revealed that the cured COPNA resin exhibited a higher favorable thermal stability than the uncured resin. A possible explanation could be that the process of curing increased the crosslinking degree of COPNA resin. The aromatic molecules were connected by the curing agent to form the 3-D structured molecules, which could lead to a better thermal stability.

Table 4 Thermogravimetric parameters of uncured and cured COPNA resins
Table 5 shows the results of the elemental analysis of the cured COPNA resin heated at different temperatures. It can be observed that the contents of O and H elements in the cured COPNA resin heated at 400 ℃ were lower than that in the case of 200 ℃, which was attributed to the removal of uncrosslinked low molecular weight compounds as mentioned above. The content of H element in the cured COPNA resin heated at 600 ℃ was much lower than that in the case of 400 ℃, which was caused by the polymerization and condensation reactions. In addition, the C/H ratio of the cured COPNA resin increased with an increasing heating temperature, which indicated that the condensation degree of the cured COPNA resin was improved gradually. The elemental analysis was consistent with the previous conclusion of TG-DTG analysis.

Table 5 Elemental compositions of cured COPNA resin heated at different temperatures
4 Conclusions
The cured COPNA resin with high thermal stability has been successfully synthesized by heating a mixture of the uncured resin with a curing agent. The curing reactionmechanism is confirmed to be electrophilic substitution reactions. Due to the cross-linking reaction between the condensed aromatic sheet and the curing agent, the cured COPNA resin with a well-ordered streamlined structure and a smooth surface showed that it had a more crystalline solid and a better molecular arrangement and orientation than the uncured resin. Thermogravimetric analyses indicated that the weight loss of the uncured and the cured COPNA resins both could be divided into three stages. The temperatures of Ti, Tfand Tmaxof the cured COPNA resin were found to be higher than those of the uncured resin. The carbon residue of the cured COPNA resin was as high as 41.65% at 600 ℃. The results show that the cured COPNA resin has preferable thermal stability and exhibits enormous potential in the application of heat resistant materials.
Acknowledgements: This work was supported by the National Natural Science Foundation of China (51172285 and 51372277); the Fundamental Research Funds for the Central Universities (14CX02060A, 15CX02084A); and the Natural Science Foundation of Shandong Province (ZR2011EL030).
[1] Otani S, Yu H A, Ota E. Carbonization behavior of condensed poly-nuclear aromatic (COPNA) resins [J]. Tanso, 1986, 127: 162-170
[2] Katsuki K, Satoru G, Shigeharu M. Carbon molecular sieving membranes derived from condensed polynuclear aromatic (COPNA) resins for gas separations [J]. Ind Eng Chem Res, 1998, 37(11): 4262-4266
[3] Ota Michiya, Otani Sugio, Iizuka Shinji, et al. Synthesis of polycondensed polynuclear aromatics (COPNA) resin from the mixture of pyrene and phenanthrene using dimethyl derivatives of benzenedimethanols as crosslinking agents [J]. Nippon Kagaku Kaishi, 1988(3): 343-350
[4] Tanemura K, Suzuki T, Nishida Y, et al. Synthesis of the strongly acidic sulfonated condensed polynuclear aromatic (S-COPNA) resins using aromatic aldehydes as crosslinking agents [J]. Polymer Bulletin, 2012, 68(3): 705-719
[5] Yan Yongli, He Li. Synthesis of condensed polynuclear aromatic resin using petroleum asphalt as monomer and its mechanism [J]. Polymer Materials Science and Engineering, 2003, 19(5): 93-96 (in Chinese)
[6] Lin Qilang, Li Tiehu. Synthesis and properties of condensed polynuclear aromatic resin using coal tar pitch as monomer and terephthalic aldehyde as cross-linking agent [J]. Polymer Materials Science and Engineering, 2007, 23(2): 62-64 (in Chinese)
[7] Guo Yansheng, Zha Qingfang, Wu Mingbo, et al. Separation of rich aromatic components from FCC slurries with extraction and synthesis of COPNA resin [J]. Journal of China University of Petroleum (Edition of Natural Science), 2002, 26(4):80-83 (in Chinese)
[8] Wu Mingbo, Shi Yangyang, Li Shibin, et al. Synthesis and characterization of condensed poly-nuclear aromatic resin derived from ethylene tar [J]. China Petroleum Processing and Petrochemical Technology, 2012, 14(4): 21-26
[9] Wu Mingbo, Jiang Wei, Wang Yuwei, et al. Synthesis of condensed polynuclear aromatic resin from furfural extract oil of reduced-pressure route II [J]. Pet Sci, 2013, 10(4): 584-588
[10] Ruan Xiangquan, Zheng Jiaming, Liu Weichang, et al. The study on mechanism of curing of the condensed polynuclear aromatic resin [J]. Journal of Tianjin University, 1997, 30(2): 217-222 (in Chinese)
[11] Ahmaruzzaman M, Sharma D K. Characterization of liquid products from the co-cracking of ternary and quaternary mixture of petroleum vacuum residue, polypropylene, Samla coal and Calotropis Procera [J]. Fuel, 2008, 87(10-11): 1967-1973
[12] Daniel M V, Uribe U N, Murgich J. Correlations between SARA fractions and physicochemical properties with1HNMR spectra of vacuum residues from Colombian crude oils [J]. Fuel, 2010, 89(1): 185-192
[13] Biswas S, Mohanty P, Sharma D K. Studies on synergism in the cracking and co-cracking of Jatropha oil, vacuum residue and high density polyethylene: Kinetic analysis [J]. Fuel Processing Technology, 2013, 106: 673-683
[14] Murakami K, Okumura M, Yamamoto M, et al. Structural analysis of mesophase pitch with high-resolution, high-temperature13C-NMR [J]. Carbon, 1996, 34(2): 187-192
[15] Shishavan R A, Ghashghaee M, Karimzadeh R. Investigation of kinetics and cracked oil structural changes in thermal cracking of Iranian vacuum residues [J]. Fuel Processing Technology, 2011, 92(12): 2226-2234
[16] Rahmani S, McCaffrey W C, Dettman H D, et al. Coking kinetics of asphaltenes as a function of chemical structure [J]. Energy & Fuels, 2003, 17(4): 1048-1056
[17] Sheremata J M, Gray M R, Dettman H D, et al. Quantitative molecular representation and sequential optimization of Athabasca asphaltenes[J]. Energy & Fuels, 2004, 18(5): 1377-1384
[18] Li Kejing, Akeredolu B A, Renehan A M, et al. Correlation of chemical and physical properties of an Alaska heavy oil from the Ugnu formation [J]. Fuel, 2013, 103: 843-849
[19] Sarkar A, Kocaefe D, Kocaefe Y, et al. Coke-pitch interactions during anode preparation [J]. Fuel, 2014, 117: 598-607
[20] Coelho R R, Hovell I, Monte M B, et al. Characterization of aliphatic chains in vacuum residues (VRs) of asphaltenes and resins using molecular modelling and FTIR techniques [J]. Fuel Processing Technology, 2006, 87(4): 325-333
[21] Liang Wenjie, Han Shuqin, Liu Wenqin. Determination of methylene and methyl in paraffinic hydrocarbons with infrared spectroscopy [J]. Journal of East China Petroleum Institute, 1987, 11(2): 86-90 (in Chinese)
[22] Andersen S I, Jensen J O, Speight J G. X-ray diffraction of subfractions of petroleum asphaltenes [J]. Energy & Fuels, 2005, 19(6): 2371-2377
[23] Siddiqui M N, Ali M F, Shirokoff J. Use of X-ray diffraction in assessing the aging pattern of asphalt fractions [J]. Fuel, 2002, 81(1): 51-58
[24] Bouhaddaa Y, Bormannb D, Sheuc E, et al. Characterization of Algerian Hassi-Messaoud asphaltene structure using Raman spectrometry and X-ray diffraction [J]. Fuel, 2007, 86(12/13): 1855-1864
[25] Prada V, Granda M, Bermejo J, et al. Preparation of novel pitches by tar air-blowing [J]. Carbon, 1999, 37: 97-106
[26] Pérez M, Granda M, García R, et al. Pyrolysis behaviour of petroleum pitches prepared at different conditions [J]. Journal of Analytical and Applied Pyrolysis, 2002, 63: 223-239
[27] Bonati A, Merusi F, Polacco G, et al. Ignitability and thermal stability of asphalt binders and mastics for flexible pavements in highway tunnels [J]. Construction and Building Materials, 2012, 37: 660-668
date: 2014-09-23; Accepted date: 2015-01-09.
Professor Zhang Zailong, E-mail: zzlyrs@upc.edu.cn; Professor Wu Mingbo, E-mail: wumb@upc. edu.cn.
杂志排行

中国炼油与石油化工的其它文章
- A Highly Efficient and Selective Water-Soluble Bimetallic Catalyst for Hydrogenation of Chloronitrobenzene to Chloroaniline
- Development of RSDS-III Technology for Ultra-Low-Sulfur Gasoline Production
- Effects of Fe2+, Co2+and Ni2+Ions on Biological Methane Production from Residual Heavy Oil
- Pyrolysis Characteristics and Kinetics of Methyl Oleate Based on TG-FTIR Method
- A Novel Thermally Coupled Reactive Distillation Column for the Hydrolysis of Methyl Acetate
- Synthesis of Waterborne Polyurethane Modified by Nano-SiO2Silicone and Properties of the WPU Coated RDX